


Fundamento y aplicabilidad de las técnicas utilizadas en el laboratorio clínico: fotometría, espectrofotometría, fluorescencia, electroforesis, cromatografía, inmunoensayos, quimioluminiscencia, química seca, nefelometría, turbidimetría.
3. Fundamento y aplicabilidad de las técnicas utilizadas en el laboratorio clínico
Las técnicas empleadas en el laboratorio clínico permiten el análisis, identificación y cuantificación de diferentes sustancias en muestras biológicas. Estas incluyen métodos como la fotometría, espectrofotometría, fluorescencia, electroforesis, cromatografía, inmunoensayos, quimioluminiscencia, química seca, nefelometría y turbidimetría.
3.1. Fotometría
Fotometría de llama
La fotometría de llama, también conocida como espectrometría de emisión atómica, se basa en el fenómeno de emisión de energía por parte de los electrones cuando pasan de un estado excitado a su estado normal. Este proceso ocurre al aplicar calor a un metal en solución, lo que provoca que sus electrones absorban energía, se exciten y, posteriormente, liberen esa energía para regresar a su estado fundamental.
La emisión de luz producida por este proceso permite identificar ciertos elementos en la muestra. Los fotómetros de llama son equipos diseñados para detectar esta emisión, especialmente en el caso de sodio y potasio. Los componentes principales de estos instrumentos son:
- Atomizador: Fragmenta la muestra en pequeñas gotas.
- Suministrador de combustible: Proporciona el gas necesario para generar la llama.
- Quemador: Donde ocurre la combustión y la excitación de los átomos.
- Monocromador: Selecciona la longitud de onda deseada para el análisis.
- Detector: Capta la radiación emitida por los átomos excitados.
- Dispositivo de lectura: Muestra los resultados del análisis.
Fundamento de la fotometría de llama
Cuando la muestra en solución es aspirada hacia la llama, el calor transforma sus componentes en átomos elementales. Estos átomos son excitados térmicamente, lo que genera un exceso de energía que posteriormente se libera en forma de luz. La intensidad de esta radiación luminosa está directamente relacionada con la cantidad de átomos que emiten energía y, por ende, con la concentración de la sustancia en la muestra.
Esta técnica es especialmente útil para la medición cuantitativa de ciertos metales.
Instrumentación y componentes de los fotómetros de llama
Los instrumentos utilizados en fotometría de llama son de doble haz y cuentan con los siguientes elementos:
- Gases para la llama:
- Se utilizan mezclas como aire-acetileno, aire-propano o hidrógeno-oxígeno.
- La temperatura generada varía según el gas empleado, lo que afecta la sensibilidad de la medición.
- Es crucial mantener una temperatura constante en la llama para garantizar la precisión. Además, se ajusta la llama para maximizar la zona interconal, ya que ahí se encuentran la mayor cantidad de átomos libres susceptibles de excitarse.
- Atomizador:
- Su función es convertir la solución en pequeñas gotas, que son transportadas hacia la llama, donde ocurren los procesos de excitación y emisión.
- Filtro o monocromador:
- Este componente selecciona la longitud de onda específica correspondiente al elemento que se desea analizar.
- Durante la combustión de la muestra, se generan energías de diferentes longitudes de onda debido a los otros componentes presentes (emisión continua). Por ello, se utiliza un monocromador con un paso de banda estrecho para eliminar las interferencias y permitir que solo llegue al detector la radiación del elemento objetivo.
- Detector:
- Generalmente son fotomultiplicadores, que amplifican la señal luminosa recibida y la convierten en datos cuantitativos.
Patrón interno en fotometría de llama
Los fotómetros de llama suelen contar con un estándar interno, generalmente litio, para garantizar la precisión de las mediciones. Este estándar ayuda a minimizar los errores provocados por fluctuaciones en la llama o variaciones en la velocidad de aspiración de la muestra. Tanto el detector que mide la muestra como el que mide el litio son afectados de forma similar por estas variaciones. Por ello, se emplea la relación entre las señales de ambos para corregir posibles errores y asegurar resultados más confiables.
Además, el litio actúa como un estabilizador de radiación. Por ejemplo, al medir potasio, la señal puede estar influida por la cantidad de sodio presente en la muestra. En ausencia de litio, la energía de los átomos excitados de sodio puede transferirse a los de potasio, lo que genera variaciones en los niveles detectados. Para evitar estos errores, las muestras se diluyen con una alta concentración de litio, lo que garantiza que la excitación del potasio sea uniforme, independientemente de la concentración de sodio.
Aplicaciones de la fotometría de llama
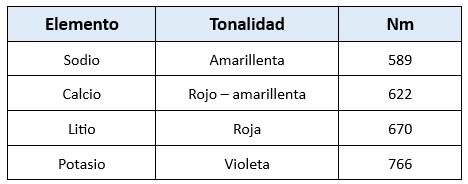
La fotometría de llama se utiliza principalmente para el análisis cuantitativo de metales alcalinos y alcalinotérreos, como sodio y potasio. Esto es posible gracias a las llamas de baja temperatura que se emplean, las cuales no excitan otros metales, evitando así interferencias.
Cada elemento genera una llama con un color característico:
- Litio: Rojo.
- Sodio: Amarillo.
- Potasio: Violeta.
- Rubidio: Rojo.
- Magnesio: Azul.
Aunque cationes como el calcio son más difíciles de excitar con llamas de baja temperatura, la sensibilidad puede mejorar utilizando llamas más calientes.
Determinaciones cuantitativas en fotometría de llama
En esta técnica, la cantidad de calor generada es limitada, lo que reduce el número de átomos excitados y, en consecuencia, las líneas espectrales obtenidas. Esto hace que las mediciones sean cuantitativas y se empleen métodos específicos para llevarlas a cabo:
1. Método directo
Aunque actualmente es menos común, este método implica la creación previa de curvas de calibración utilizando soluciones de concentraciones conocidas. Los valores obtenidos de la muestra se extrapolan a estas curvas para determinar la concentración del compuesto analizado.
2. Método de adición del patrón
Este método utiliza como patrón el mismo elemento que se busca medir. El equipo se calibra a cero utilizando un patrón en blanco y a 100 con una muestra de alta concentración. Los valores obtenidos se extrapolan en una línea construida con estos datos.
- Ventaja principal: Al usar el mismo elemento como patrón, se eliminan las interferencias que podrían alterar los resultados.
3. Método del patrón interno
En este caso, se utiliza como patrón un elemento con propiedades similares al que se desea medir, pero que no esté presente en la muestra. Para muestras biológicas, el litio es la elección más común, ya que tiene una emisión característica de 671 nm. Este método es especialmente útil para obtener mediciones precisas y evitar interferencias de otros componentes presentes en la muestra.
3.2. Espectrofotometría
Consideraciones generales
La espectrofotometría comprende un conjunto de técnicas instrumentales que se basan en la interacción de la energía con la materia. Este tipo de interacción puede manifestarse a través de fenómenos como absorción, emisión, transmisión o dispersión.
¿Sabías que…?
La espectrofotometría es un método de análisis químico cuantitativo que utiliza la luz para determinar la concentración de sustancias químicas. Dependiendo de la radiación empleada, puede clasificarse como espectrofotometría de absorción visible (también conocida como colorimetría), ultravioleta o infrarroja.
Naturaleza de la energía radiante
La energía radiante se propaga como radiación electromagnética (REM), que presenta un comportamiento dual, es decir, actúa como una onda en fenómenos como refracción y difracción, y como partículas (fotones) en procesos de absorción y emisión. Este principio es conocido como la Dualidad Onda-Corpúsculo.
Cuando se considera la REM como una onda, existen ciertos parámetros característicos:
1. Longitud de onda (λ): Es la distancia entre dos puntos equivalentes de una onda. Se mide en unidades de longitud, como el angstrom (Å), donde:
1 Å = 0,1 milimicras (mμ) = 10-10m.
La inversa de la longitud de onda () se denomina número de onda (ν), que se mide en cm-1 y está relacionado con la frecuencia.

Espectro electromagnético
El espectro electromagnético abarca un amplio rango de frecuencias y longitudes de onda. Las radiaciones con menor longitud de onda y mayor frecuencia poseen mayor energía, mientras que aquellas con mayor longitud de onda y menor frecuencia tienen menos energía. Esto se relaciona directamente con la ecuación de Planck.
- Radiaciones de alta energía (como los rayos cósmicos y gamma): Estas pueden inducir transformaciones nucleares en la materia.
- Rayos X: Provocan transiciones de electrones en las capas internas de los átomos.
- Radiación ultravioleta-visible (UV-VIS): Produce transiciones electrónicas en las capas externas de los átomos, pudiendo romper enlaces químicos y originar nuevos compuestos. Estas transiciones son la base de las técnicas espectroscópicas UV-VIS.
- Radiación infrarroja (IR): Genera cambios en las vibraciones moleculares, base de la espectroscopia infrarroja.
- Microondas: Alteran la rotación molecular.
- Ondas de radio: Producen cambios en los momentos magnéticos de los átomos, fundamento de la resonancia magnética nuclear (RMN).
Fenómenos de interacción entre luz y materia
Cuando la luz interactúa con la materia, pueden producirse fenómenos como la absorción y la emisión de energía.
1. Proceso de absorción
Cuando una partícula en estado fundamental (reposo) entra en contacto con un haz de luz, absorbe su energía y pasa a un estado excitado. Este cambio depende de las características de la partícula (como sus enlaces) y de la energía o longitud de onda de la luz que interactúa. Posteriormente, la partícula regresa de manera espontánea a su estado fundamental, liberando la energía absorbida en forma de calor. Este proceso se representa como:
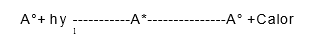
Donde:

Fundamentos de la espectrofotometría
La espectrofotometría se basa en los principios descritos anteriormente. Dependiendo del enfoque, las técnicas pueden centrarse en medir la absorción de energía emitida por una fuente (espectrofotometría de absorción) o en medir la energía emitida por los compuestos tras ser excitados (espectrofotometría de emisión).
Espectrofotometría de absorción
A) Fundamento
La espectrofotometría de absorción consiste en medir la cantidad de luz que llega al detector después de haber sido absorbida por una sustancia. En química clínica, las mediciones suelen realizarse dentro del rango de longitudes de onda de 220 a 800 nm.
Aunque el término “determinaciones espectrofotométricas” hace referencia a la medición de la intensidad de luz en un espectro más estrecho que la fotometría, actualmente la principal diferencia entre un fotómetro y un espectrofotómetro radica en el método utilizado para seleccionar la longitud de onda.
- Los fotómetros o colorímetros emplean filtros para seleccionar la longitud de onda.
- Los espectrofotómetros utilizan prismas o redes de difracción para este propósito.
B) Leyes de absorción
Cuando un haz de luz de intensidad inicial (Io) pasa a través de una solución contenida en una cubeta, la sustancia absorbente reduce la intensidad del haz saliente (Is) debido a la absorción de luz.
La transmitancia se define como la relación entre la luz que sale de la solución y la que entra, y se expresa como:
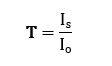
En términos porcentuales, la transmitancia es el porcentaje de luz que atraviesa la solución:
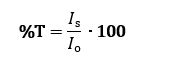
Por otro lado, la absorbancia (A) mide la cantidad de luz que ha sido absorbida y depende directamente de la concentración de la sustancia absorbente en la solución.
Para garantizar que la absorción medida corresponde únicamente al compuesto de interés, se utiliza una solución de referencia o blanco que contiene todos los componentes excepto la sustancia que se quiere analizar. Las mediciones posteriores se comparan con esta referencia, utilizando la misma cubeta para evitar variaciones.
Aunque hoy en día la transmitancia directa (0% T) se emplea con menor frecuencia, es importante recordar que lo que se mide realmente es la luz transmitida, y la absorbancia se calcula a partir de esta. A su vez, la concentración del compuesto se deduce de la absorbancia.
Sabías que…
La transmitancia es un parámetro que indica la diferencia entre la cantidad de luz que atraviesa una solución coloreada y la que emerge después de haber pasado a través de un determinado volumen de dicha disolución.
- Se expresa en porcentaje de luz.
- Representa la relación entre la luz incidente y la luz transmitida.
La Ley de Beer describe cómo la absorbancia de una solución está vinculada a dos factores: la concentración de la sustancia absorbente y la distancia que recorre el haz de luz dentro de la solución. De acuerdo con esta ley, la absorbancia es directamente proporcional a la concentración del soluto y a la longitud del trayecto que la luz atraviesa.
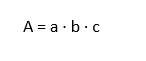
diciendo:
- A = absorbancia.
- a = coeficiente de absorción.
- b = distancia recorrida por la luz en centímetros.
- c = concentración del absorbente.
Esta ecuación es la base fundamental del análisis cuantitativo en los estudios espectrofotométricos.
La absorbancia es una magnitud adimensional, es decir, no tiene unidades. En cambio, el coeficiente de absorción es una constante que depende de la naturaleza química del soluto y cuyas unidades son inversamente proporcionales a las de a, b y c .
El coeficiente de absorción, también llamado coeficiente de extinción molar y representado con el símbolo ε , se define cuando la longitud del paso de luz ( b ) se mide en centímetros y la concentración ( c ) en moles por litro. Este coeficiente es específico para cada compuesto y permanece constante si las condiciones de la medición, como la longitud de onda, el pH, el disolvente y la temperatura, se mantienen sin cambios. Su unidad de medida es 1/mol·cm .
Cuanto mayor sea ε para una determinada sustancia en condiciones fijas de trabajo, mayor será la absorbancia de dicha sustancia en esas mismas condiciones, lo que aumenta la sensibilidad del método utilizado para su detección.
Métodos para determinar la concentración de una sustancia.
La Ley de Beer permite aplicar diferentes métodos experimentales para determinar la concentración de un compuesto en solución. Basándonos en la relación lineal entre absorbancia y concentración, existen dos maneras de obtener la concentración de una muestra desconocida:
a) Comparación con una solución de referencia :
Si se dispone de una solución problema ( P ) y otra de concentración conocida ( S ), se establece la siguiente relación matemática entre ellas:
As = absorbancia de la solución estándar.
Ap = absorbancia de la solución problema.
Cs = concentración de la solución estándar.
Cp = concentración de la solución problema.
Curva de calibración :
Consiste en la representación gráfica de la absorbancia (eje vertical) en función de la concentración (eje horizontal). Para construir la curva de calibración, se miden las absorbancias de varias soluciones de concentración conocida y se grafican estos valores, obteniendo una línea recta. Posteriormente, al medir la absorbancia de la muestra problema, su concentración se determina mediante interpolación en la gráfica.
Es importante destacar que estos métodos solo son aplicables si la sustancia analizada cumple con la Ley de Beer , es decir, si la relación entre absorbancia y concentración es lineal. Además, las soluciones problema y estándar deben ser analizadas bajo las mismas condiciones experimentales.
Desviaciones de la Ley de Beer
Existen situaciones en las que la relación entre absorbancia y concentración deja de ser lineal, generando desviaciones en la Ley de Beer . Estas desviaciones pueden deberse a diversos factores, tales como:
- Uso de concentraciones demasiado elevadas del compuesto analizado.
- Empleo de radiación no monocromática.
- Absorbancia significativa del disolvente en comparación con la del soluto.
- Presencia de luz dispersa o errática que afecta la medición.
- Irregularidades en la cubeta de medición, como caras no paralelas.
- Interferencias provocadas por otras sustancias que absorben en la misma longitud de onda.
- Fenómenos como la fluorescencia.
Componentes de un espectrofotómetro
Los dispositivos utilizados para medir la absorción de luz en una muestra, conocidos como espectrofotómetros de absorción , están compuestos por cinco elementos esenciales:
1. Fuente de radiación : Emite la luz utilizada para realizar la medición.
2. Selector de longitud de onda : Permite aislar la radiación de la longitud de onda específica que se desea analizar.
3. Destinatario para la muestra : Generalmente una cubeta de vidrio o plástico donde se coloca la solución a estudiar.
4. Detector de radiación o transductor : Captura la luz transmitida y la convierte en una señal eléctrica.
5. Registrador o medidor : Permite visualizar y cuantificar los valores obtenidos en la medición.
1. Fuente de radiación
Este es el dispositivo encargado de generar la luz que se estudiará en el análisis. Se distinguen dos tipos:
- Fuentes continuas: Producen una radiación compuesta por una amplia variedad de longitudes de onda. Se utilizan en la absorción molecular.
- Para el rango Ultravioleta (UV), se emplean lámparas de Deuterio y de hidrógeno, mientras que en el Visible se usa la lámpara de filamento de tungsteno.
- Cuando se requiere una fuente de luz con alta intensidad, se opta por lámparas de arco llenas de gas (como argón, xenón o mercurio) operando a alta presión.
- En el caso del Infrarrojo (IR), se utilizan materiales sólidos inertes que se calientan eléctricamente a temperaturas entre 1200 y 2000 °K, generando así una radiación continua.
- Fuentes discontinuas: Emiten radiaciones en forma de unas pocas líneas correspondientes a longitudes de onda específicas. Son empleadas en técnicas como la absorción atómica, fluorescencia y Raman.
- Un caso especial de fuentes discontinuas en la composición de los láseres, que se utilizan por su alta intensidad, estrecho ancho de banda y la coherencia de la luz que producen.
2. Selector de longitud de onda
Este componente se encarga de separar la radiación policromática emitida por la fuente en bandas que cubren un rango limitado de longitudes de onda. Se presentan dos variantes:
- Monocromadores: Permiten ajustar la longitud de onda seleccionada mediante el uso de un prisma o una rejilla de difracción, que dispersa la radiación en sus componentes individuales.
- Los monocromadores basados en prismas, fabricados en materiales como CINa, vidrio o cuarzo, son cada vez menos utilizados. La refracción entre sus caras genera la dispersión angular de la luz.
- Por otro lado, los monocromadores que emplean redes de difracción son más económicos y ofrecen una separación superior de las longitudes de onda, ya que dispersan la radiación de forma lineal. En estas redes, pequeñas hendiduras ubicadas en un ángulo específico actúan como mini prismas. Generalmente se prefieren las rejillas de reflexión porque minimizan la pérdida de energía.
- Filtros: Se divide en filtros de absorción y de interferencia.
- Filtros de absorción: Funcionan limitando la radiación al absorber ciertos segmentos del espectro. Suelen consistir en vidrios coloreados o en colorantes dispersos en gelatina, montados entre dos placas de vidrio. Son económicos, pero su aplicación se restringe al espectro visible. Un filtro de color absorbe específicamente su propio color, dejando pasar las longitudes de onda correspondientes a los otros dos colores primarios (el color complementario). Por ello, el filtro seleccionado será de color complementario al de la muestra analizada.
- Filtros de interferencia: Se utilizan en los rangos UV-VIS (ultravioleta-visible) e IR (infrarrojo) y están compuestos por delgadas películas semitransparentes metálicas depositadas en cada cara de un material dieléctrico, como el fluoruro cálcico o magnésico.
3. Destinatario de la muestra
Con la excepción de la espectroscopia de emisión, la mayoría de los análisis requieren celdas o cubetas para contener la muestra. Estas deben estar fabricadas con un material que sea transparente en la región del espectro en la que se trabaja.
- Para mediciones en UV, se requieren cubetas de cuarzo o sílice fundido, las cuales también son aptas para algunas aplicaciones en el VIS y parte del IR.
- En el rango visible, se pueden utilizar cubetas de vidrio o plástico.
- En el IR, se prefiere el cloruro sódico cristalizado.
Es fundamental que la cubeta esté perfectamente limpia, sin rayas, aristas o burbujas, para garantizar resultados precisos y reproducibles.
4. Detector
Después de que la radiación electromagnética (REM) ha atravesado la cubeta, una parte de ella es absorbida por el componente analizado y otra parte es transmitida. El detector es el dispositivo que convierte esta transmisión REM en una señal eléctrica. Se clasifican en dos tipos:
- Detectores de fotones: Pueden consistir en células fotovoltaicas, células de capa de barrera o fotocélulas; fototubos o tubos fotomultiplicadores. En cada caso, la luz que incide sobre el detector provoca la liberación de electrones, generando una corriente eléctrica proporcional a la REM recibida.
- Detectores térmicos: Las radiaciones de baja energía, como las del infrarrojo, no generan la emisión de electrones necesaria en los detectores de fotones. Por ello, se utiliza la detección basada en el incremento de temperatura (fotoconducción), que se puede medir con diferentes dispositivos, tales como termocúpulas, bolómetros o el detector neumático de Golay.
5. Sistema de procesamiento de señales
Este sistema se encarga de interpretar la señal eléctrica emitida por el detector y convertirla en un valor final, que puede ser transmitancia, absorbancia o, en ocasiones, concentración. La señal del detector se puede mostrar mediante el movimiento de una aguja en una escala oa través de una representación digital en una pantalla. Aunque el detector siempre mide la transmitancia, el sistema de procesamiento se encarga de convertirla en otros parámetros, según sea necesario para el análisis.
D) Aspectos prácticos
A) Uso de blancos
Antes de realizar una medición, es fundamental emplear un blanco para eliminar cualquier posible interferencia causada por la absorción o reflexión de la cubeta o del disolvente en el que está disuelta la sustancia de interés. En otras palabras, el blanco permite establecer un punto de referencia al asignar un valor “cero” a todo aquello que puede influir en la medición, pero que no forma parte de la muestra en sí, sino del material o los reactivos utilizados.
La metodología consiste en realizar una primera medición con la cubeta que solo contiene el disolvente. Esto permite establecer una intensidad inicial que servirá como referencia, asegurando que cualquier absorción adicional sea atribuida únicamente a la muestra en análisis. Posteriormente, la absorbancia medida en el blanco se resta de las absorbancias obtenidas en las mediciones de las muestras problema.
Existen distintos tipos de blancos:
- Blanco de suero: Se realiza una medición inicial con el suero diluido en la misma proporción en un disolvente inerte, con el que no reaccione. Esto permite eliminar posibles interferencias causadas por la propia muestra, como la presencia de hemólisis, lipemia o bilirrubinemia antes de que se lleve a cabo la reacción.
- Blanco de reactivos: Su objetivo es corregir interferencias originadas por los reactivos empleados en el análisis. Para ello, se efectúa una primera medición utilizando únicamente el reactivo, antes de añadir la muestra.
B) Medición en múltiples longitudes de onda
Interferencias cinéticas
Estas interferencias se deben a la presencia de sustancias en la muestra que pueden reaccionar con alguno de los componentes de la mezcla analítica. La relevancia de estas interferencias depende tanto del tiempo de reacción como de la concentración de las sustancias involucradas.
Si la sustancia interferente reacciona a una velocidad superior a la del compuesto que se analiza, el problema puede corregirse utilizando un análisis cinético. En cambio, si ambas reaccionan a la misma velocidad, la corrección se realiza mediante la comparación de los resultados obtenidos en presencia y en ausencia de la sustancia interferente, aplicando una simple sustracción.
Determinación de sustratos mediante absorción molecular
Como se ha mencionado anteriormente, la relación entre la absorbancia de una solución y la concentración de la sustancia en cuestión se establece a través de la Ley de Lambert-Beer.
Para cuantificar la concentración de un compuesto en solución, se mide su absorbancia. Si el compuesto no absorbe luz en la región espectral de interés, se puede inducir un cambio químico para transformarlo en otro compuesto que sí lo haga. Esto se logra mediante la reacción del analito con reactivos específicos o integrándolo en una serie de reacciones químicas que generen un producto con propiedades de absorción adecuadas.
En estos casos, la reacción que permite la conversión del analito en un compuesto absorbente se denomina reacción auxiliar, mientras que la reacción utilizada para la medición recibe el nombre de reacción indicadora. Ambas pueden ser simples reacciones químicas o procesos catalizados por enzimas.
Métodos espectrofotométricos para medir concentraciones en solución
Existen dos enfoques principales en espectrofotometría de absorción molecular para la cuantificación de sustancias en solución:
1. Método de punto final o medición en dos puntos
En este método, la muestra se incuba junto con los reactivos y un estándar de concentración conocida durante un tiempo determinado, hasta que la reacción se completa o se alcanza el equilibrio. Este procedimiento es más apropiado llamarlo método de equilibrio, ya que en algunas ocasiones es posible realizar la medición antes de llegar al punto final de la reacción.
Cuando se utilizan enzimas como reactivos en este tipo de mediciones, se añade un exceso de sustrato y se deja reaccionar con la muestra durante un tiempo definido. Durante este proceso, la transformación del sustrato en producto provoca un cambio en la absorbancia (aumento o disminución), el cual es proporcional a la cantidad de enzima presente en la muestra. En estos casos, se puede cuantificar tanto la cantidad de sustrato que no ha reaccionado como la cantidad de producto generado.
2. Método cinético
Este método se basa en la medición de la velocidad de reacción, es decir, el cambio en la absorbancia a lo largo del tiempo. La actividad enzimática o la concentración del analito se determinan a partir de ecuaciones derivadas de la Ley de Lambert-Beer.
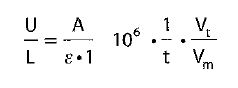
Siendo:
- AA = variación de absorbancia.
- t = tiempo de reacción.
- ε = coeficiente de extinción molar.
- l = longitud del paso de la cubeta.
- Vt = volumen total de la reacción.
- Vm = volumen de la muestra.
- 10⁶ = factor de conversión de unidades.
Tipos de espectrofotometría de absorción
Los sistemas basados en espectrofotometría de absorción pueden clasificarse de acuerdo con el tipo de energía luminosa emitida por la fuente. Entre ellos, se destaca la espectroscopia de absorción ultravioleta-visible (UV-VIS), la cual se divide en dos regiones:
- Región ultravioleta (UV): Comprende las longitudes de onda entre 195 y 380 nm.
- Región visible (VIS): Incluye el rango de longitudes de onda perceptible por el ojo humano, entre 390 y 780 nm. Los colores ordenados de mayor a menor longitud de onda son:
Violeta > Azul > Verde > Amarillo > Naranja > Rojo.
Dado que la energía de la radiación es inversamente proporcional a su longitud de onda, la radiación ultravioleta es más energética que la luz visible (a mayor λ, menor energía).
A) Principios básicos
La absorción de este tipo de radiación ocurre cuando los electrones externos de los átomos se excitan al absorber energía. Este fenómeno es particularmente relevante en los compuestos orgánicos, aunque algunos aniones inorgánicos, como los carbonatos y nitritos, también pueden absorber en esta región del espectro.
Para que una molécula pueda absorber radiación en el rango UV-VIS, debe contener grupos cromóforos, que son estructuras funcionales con enlaces dobles o triples, así como sistemas de dobles enlaces conjugados. Algunos ejemplos de estos grupos son los alquenos, alquinos, carbonilo, carboxilo y amido, entre otros.
Además de los cromóforos, existen otros grupos llamados auxocromos, que no absorben luz por sí mismos, pero pueden influir en la absorción del cromóforo al que están unidos. Los auxocromos suelen ser grupos funcionales que contienen átomos con pares de electrones no compartidos, como oxígeno, halógenos, azufre y nitrógeno.
Cuando se representa la absorbancia a distintas longitudes de onda, se obtiene una curva característica denominada espectro de absorción. Este espectro puede verse afectado por varios factores, entre ellos:
- La presencia de auxocromos.
- Cambios en el pH de la solución.
- Variaciones en la concentración salina del medio en el que se encuentra la muestra.
Efectos sobre el espectro de absorción
Dependiendo de las condiciones experimentales y de la estructura química del compuesto, el espectro de absorción puede sufrir modificaciones, dando lugar a diferentes efectos:
- Efecto hipsocrómico: Desplazamiento del espectro hacia menores longitudes de onda (mayor frecuencia).
- Efecto batocrómico: Desplazamiento del espectro hacia longitudes de onda mayores (menor frecuencia).
- Efecto hipercrómico: Aumento en la intensidad de absorción.
- Efecto hipocrómico: Disminución en la intensidad de absorción.
Sabías que…
La espectrofotometría UV-VIS se basa en la absorción de fotones en la región de la radiación ultravioleta y visible. Este método emplea luz en el espectro visible y en regiones cercanas, como el ultravioleta cercano (UV) y el infrarrojo cercano (IR). En este rango del espectro electromagnético, las moléculas experimentan transiciones electrónicas.
Además, esta técnica es complementaria a la espectrometría de fluorescencia, que analiza las transiciones desde un estado excitado hacia el estado basal, mientras que la espectrofotometría de absorción mide las transiciones desde el estado basal al estado excitado.
B) Instrumentación
1. Fuente de radiación
Las fuentes de radiación en espectroscopia suelen ser lámparas que emiten radiación continua, basándose en el fenómeno de emisión del cuerpo negro. Esta propiedad describe la capacidad de los cuerpos sólidos para emitir radiación cuando son sometidos a temperaturas elevadas. En este proceso, la longitud de onda de la radiación emitida no depende del material del sólido, sino únicamente de la temperatura a la que se encuentra.
Algunas de las fuentes de radiación más utilizadas en espectroscopia infrarroja incluyen:
- Lámpara de Nernst
- Lámpara de Globar
- Lámpara de filamento de níquel y cromo
2. Selector de longitud de onda
Para seleccionar la longitud de onda deseada dentro del espectro de la radiación emitida, se emplean los siguientes dispositivos:
- Monocromadores:
- Prismas: Son fabricados con materiales que permiten el paso de la radiación infrarroja, como los haluros de metales alcalinos y alcalinotérreos (KBr, NaCl, LiF, CaF₂, etc.).
- Sin embargo, estos materiales son higroscópicos, es decir, tienden a absorber la humedad del ambiente, lo que puede afectar su funcionamiento. Por esta razón, deben ser almacenados en condiciones secas y protegidas.
- Redes de difracción: Se utilizan principalmente rejillas reflectantes de aluminio, que presentan una mayor resistencia a la humedad y ofrecen un mejor desempeño en la dispersión de la luz.
- Prismas: Son fabricados con materiales que permiten el paso de la radiación infrarroja, como los haluros de metales alcalinos y alcalinotérreos (KBr, NaCl, LiF, CaF₂, etc.).
- Filtros: También es posible utilizar filtros de interferencia, los cuales permiten aislar rangos específicos de longitudes de onda.
3. Célula de muestra (cubeta)
La espectroscopia infrarroja permite analizar muestras en diferentes estados de agregación: sólidos, líquidos y gases. No existe un disolvente completamente transparente a la radiación infrarroja, por lo que la preparación de la muestra debe realizarse con cuidado.
- Muestras sólidas:
- Análisis en suspensión: Se dispersa el sólido en un medio que no absorba la radiación infrarroja, generalmente un polímero de alto peso molecular. La suspensión se coloca entre dos ventanas de cloruro de sodio (NaCl).
- Análisis en pastilla: Se pulveriza la muestra sólida y se mezcla con bromuro de potasio (KBr). Luego, se prensa hasta formar una pastilla que se empleará como célula de muestra.
- Muestras líquidas: Se coloca una pequeña cantidad del líquido entre dos discos de material transparente, como cloruro de sodio (NaCl).
- Muestras gaseosas: Se introducen en una cubeta con superficies internas reflectantes, lo que permite aumentar la trayectoria de la luz dentro de la muestra, mejorando así la sensibilidad del análisis.
4. Detector
Dado que la radiación infrarroja posee una menor intensidad y energía en comparación con otras regiones del espectro electromagnético, los detectores empleados en esta técnica deben ser detectores térmicos, los cuales operan en base al principio de termoconducción.
Entre los detectores más utilizados en espectroscopia infrarroja se encuentran:
- Termocúpulas
- Bolómetros
- Detector neumático de Golay
De manera similar a la espectroscopia UV-VIS, los instrumentos pueden operar bajo dos configuraciones principales:
- Instrumentos de haz simple: Donde la medición se realiza secuencialmente, primero con la muestra de referencia y luego con la muestra problema.
- Instrumentos de doble haz: En los que la radiación se divide en dos caminos, permitiendo medir simultáneamente la muestra de referencia y la muestra problema, lo que mejora la estabilidad y precisión de la medición.
C) Aplicaciones
1. Análisis cuantitativo
El uso de la espectroscopia infrarroja para cuantificación presenta ciertas limitaciones instrumentales y espectrales. Además, en esta región del espectro, la Ley de Lambert-Beer tiende a presentar desviaciones, lo que dificulta su aplicación en mediciones exactas de concentración. Por esta razón, esta técnica no es ampliamente utilizada para análisis cuantitativos.
2. Análisis cualitativo
La espectroscopia infrarroja se emplea principalmente para identificar compuestos orgánicos, ya que permite detectar la presencia de grupos funcionales, los cuales presentan frecuencias de absorción características dentro del espectro IR.
Espectroscopía de Absorción Atómica
A) Fundamento
En esta técnica, el elemento que se desea analizar se separa de sus enlaces químicos y se encuentra en estado atómico neutro, sin excitación ni ionización. En este estado de baja energía, los átomos pueden absorber radiación en un rango de longitudes de onda muy estrecho (0,001 – 0,01 nm).
La cantidad de energía absorbida por los átomos es equivalente a la que emitirían si se encontraran en estado excitado. Se puede realizar una corrección basada en la Ley de Lambert-Beer.
A diferencia de los métodos de emisión, donde solo una pequeña fracción de los átomos se excita, en la espectroscopía de absorción atómica la mayoría de los átomos permanecen en estado neutro y pueden absorber radiación, lo que hace que esta técnica sea altamente sensible para la detección de elementos.
B) Instrumentación
1. Fuente de radiación
Las fuentes de radiación utilizadas en espectroscopia de absorción atómica pueden ser de dos tipos:
- Lámparas de cátodo hueco
- Lámparas de descarga sin electrodos
Lámpara de cátodo hueco
- Diseñada para emitir luz en una longitud de onda específica correspondiente al metal que se desea analizar.
- Contiene un cátodo hecho del metal en estudio, aunque en algunos casos puede ser una aleación de varios elementos.
- El gas de relleno de la lámpara suele ser neón o argón.
- Se aplica una corriente entre los electrodos, lo que provoca la emisión de átomos metálicos desde el cátodo hacia las paredes internas de la lámpara.
- Cuando estos átomos colisionan con el gas de relleno, pierden energía y emiten una radiación característica del metal analizado.
- Por ejemplo, en la detección de calcio, solo los átomos de calcio absorberán la luz específica emitida por la lámpara de cátodo hueco cuando atraviese la muestra.
Lámpara de descarga sin electrodos
- Este tipo de fuente es más intensa que la lámpara de cátodo hueco.
- Está compuesta por un tubo de cuarzo, que contiene una pequeña cantidad del metal o de su sal junto con un gas inerte (como argón).
- En lugar de utilizar electrodos, la excitación se lleva a cabo mediante radiación de microondas o radiofrecuencia.
- El gas inerte se ioniza y los iones resultantes se aceleran hasta alcanzar la energía necesaria para excitar los átomos del metal en análisis, lo que genera su radiación característica.
Sabías que…
Si se necesita una fuente de radiación de alta intensidad en espectrofotometría de absorción atómica, se recomienda el uso de lámparas de cátodo hueco rellenas de gas o lámparas de descarga sin electrodos.
Espectroscopía de Emisión
Los sistemas de espectroscopía de emisión permiten detectar sustancias mediante la energía que estas liberan después de haber sido estimuladas o excitadas por una fuente de radiación.
A) Espectroscopía de Emisión de Fluorescencia
1. Fundamento
La espectroscopía de fluorescencia es una técnica basada en la emisión de luz por moléculas que han absorbido radiación, generalmente en la región ultravioleta (UV). Cuando una molécula absorbe esta radiación, pasa de su estado fundamental a un estado excitado con mayor energía. Al regresar a su estado original, una parte de la energía absorbida se disipa en forma de calor, mientras que otra parte se emite como radiación fluorescente.
Esta radiación emitida tiene menor energía que la absorbida, lo que significa que su longitud de onda es mayor. La diferencia entre la longitud de onda de excitación y la de emisión se conoce como el desplazamiento de Stokes. Este proceso ocurre en aproximadamente 10⁻⁸ segundos, es decir, casi simultáneamente con la absorción de la radiación.
Para que una sustancia sea fluorescente, debe contener electrones capaces de excitarse, lo cual es común en ciertos tipos de moléculas:
- Moléculas orgánicas fluorescentes: aquellas con múltiples enlaces dobles conjugados, estructuras rígidas y multicíclicas, como los nucleótidos, esteroides y algunas vitaminas.
- Moléculas inorgánicas fluorescentes: suelen ser metales que forman quelatos.
La relación entre la intensidad de la radiación fluorescente y la concentración del compuesto sigue la Ley de Lambert-Beer:
F = K · Φ · I₀ · (ε · l · c)
F = Intensidad de la radiación fluorescente recibida por el detector
Donde:
- I₀ = Intensidad de la radiación incidente.
- Φ = Eficiencia cuántica (fracción de moléculas excitadas que emiten luz).
- K = Constante dependiente del instrumento.
- ε = Coeficiente de extinción molar.
- l = Longitud del paso de luz en la cubeta.
- c = Concentración del compuesto fluorescente.
Dado que la fluorescencia es proporcional a la intensidad de la luz incidente, se requieren fuentes de radiación de alta intensidad para obtener una buena señal. Esta técnica es especialmente útil para analizar soluciones diluidas, ya que su límite de detección es más bajo en comparación con la espectroscopía UV-VIS.
2. Instrumentación
- Fuente de radiación: Debe proporcionar alta intensidad. Se pueden usar:
- Lámpara de mercurio (emisión discontinua).
- Lámpara de arco de xenón (emisión continua).
- Selector de longitud de onda: Se requieren dos selectores, uno para la excitación y otro para la emisión. Se pueden usar:
- Filtros: de vidrio, cuarzo o interferenciales. Si un fluorómetro emplea filtros, se denomina fluorómetro de filtros.
- Monocromadores: si el sistema emplea monocromadores, se llama espectrofluorómetro.
- Prismas de cuarzo y redes de difracción.
- Cubetas: Similares a las utilizadas en la espectroscopía UV-VIS, pueden ser de cuarzo o vidrio pirex.
- Detector: Normalmente se utilizan tubos fotomultiplicadores. Dado que la radiación fluorescente se emite en múltiples direcciones, se selecciona un ángulo adecuado para evitar interferencias con la radiación incidente.
3. Tipos de Instrumentos
- Fluorómetros de filtro.
- Espectrofotómetros de fluorescencia, que pueden ser:
- De haz sencillo.
- De doble haz.
4. Aplicaciones
Las sustancias fluorescentes pueden emplearse como marcadores en fluoroinmunoanálisis (ej. fluoresceína, rodamina, umbeliferona). También se pueden detectar compuestos no fluorescentes haciéndolos reaccionar con reactivos que los transformen en fluorescentes.
Esta técnica permite la identificación de compuestos como nucleótidos (NAD, ATP, NADP), enzimas, esteroides y vitaminas (B6, A, E).
- Análisis Cuantitativo: Es útil para detectar elementos en trazas, dado su bajo límite de detección.
- Análisis Cualitativo: Se compara el espectro de excitación y emisión de la muestra con los espectros de referencia de la sustancia sospechada.
B) Espectroscopía de Emisión Atómica
Esta técnica emplea fuentes de excitación con mayor energía que una llama, lo que permite obtener espectros más detallados con múltiples líneas de emisión.
1. Instrumentación
- Fuente de excitación: La elección de la fuente influye en el tipo e intensidad de las líneas espectrales. Algunas opciones incluyen:
- Arco y chispa: Excitación entre electrodos, adecuada para muestras líquidas o sólidas y permite analizar varios elementos simultáneamente.
- Láser.
- Plasma de argón: Es una mezcla gaseosa donde una fracción de los átomos o moléculas está ionizada. Cuando la muestra se introduce en este medio, se atomiza debido a la alta temperatura.
- Detectores: Se emplean películas fotográficas o tubos fotomultiplicadores (estos últimos son más costosos).
2. Aplicaciones
Principalmente se usa en análisis cualitativo para la identificación de elementos en una muestra, utilizando detectores fotográficos.
3.3. Fluorescencia
Se denomina fluorescencia a la capacidad que tienen ciertas sustancias (llamadas fluorescentes) de emitir radiación de mayor longitud de onda tras ser expuestas a radiaciones de onda corta.
La intensidad de la fluorescencia es proporcional a la radiación incidente. Por esta razón, el microscopio de fluorescencia cuenta con una fuente de luz intensa y un filtro para radiación ultravioleta (UV). La luz incide sobre la muestra y, si contiene sustancias fluorescentes, estas emitirán luz visible. Este tipo de microscopio incluye un filtro de excitación y otro de emisión.
La microscopía de fluorescencia se clasifica en:
- Primaria: cuando la muestra presenta fluorescencia de manera natural.
- Secundaria: cuando la muestra ha sido marcada con colorantes fluorescentes.
- Inmunofluorescencia: cuando el colorante fluorescente está unido a un anticuerpo marcado.
Algunas sustancias fluorescentes comúnmente utilizadas en microscopía son:
- Auramina: empleada en la detección del bacilo de la tuberculosis, ya que bajo luz UV emite fluorescencia, permitiendo su identificación.
- Naranja de acridina.
- Fluoresceína y rodamina: se combinan con anticuerpos marcados para evidenciar antígenos.
- Antígenos marcados: se utilizan para detectar anticuerpos.
3.4. Electroforesis
Introducción
La electroforesis es un fenómeno electrocinético que implica el movimiento de partículas cargadas dentro de una solución cuando se aplica un campo eléctrico.
Este método permite la separación de moléculas cargadas eléctricamente según su velocidad de migración en un campo eléctrico, el cual se genera mediante electrodos sumergidos en la solución. Las moléculas con carga positiva se desplazan hacia el cátodo, mientras que las de carga negativa migran hacia el ánodo.
Factores que influyen en la movilidad electroforética
- Voltaje aplicado: un voltaje mayor mejora la separación de moléculas.
- Carga de la partícula: la movilidad depende del pH, por lo que se emplean soluciones tamponadas para mantenerlo estable.
- Coeficiente de fricción: está determinado por la viscosidad del disolvente, así como por la forma y el tamaño de las partículas.
- Fuerza iónica del tampón: su incremento aumenta la corriente transportada y reduce la velocidad de migración. Esta fuerza varía con la molaridad y la carga del ión.
- Medio de soporte: influye si posee cargas.
- Tiempo de duración: como la velocidad de migración es constante, un mayor tiempo de aplicación implica un mayor desplazamiento.
- Temperatura: también puede afectar la movilidad.
Materiales y procedimiento
El equipo de electroforesis incluye un recipiente con electrodos (ánodo y cátodo), un soporte de electroforesis en contacto con una solución tampón y una fuente de alimentación de corriente que permite programar la intensidad o el voltaje del campo eléctrico.
El procedimiento básico consiste en depositar la muestra sobre el soporte, aplicar corriente durante el tiempo necesario para que las moléculas se separen y, posteriormente, revelar las fracciones obtenidas. Estas pueden cuantificarse mediante un densitómetro (tras transparentación) o por espectrofotometría UV-VIS tras diluir y procesar las manchas.
Los soportes varían según la aplicación:
- Papel: existen diferentes tipos, pero su uso es limitado. El papel de filtro se usa raramente (por ejemplo, en la separación de aminoácidos). El acetato de celulosa permite una migración rápida y una mayor resolución, por lo que se emplea ampliamente en la separación de proteínas (proteinograma), que se revelan con rojo Ponceau.
- Agarosa: se prefiere al agar, ya que minimiza la electroósmosis y la adsorción, lo que la hace ideal para técnicas inmunológicas y la separación de ácidos nucleicos.
- Gel de poliacrilamida: es el soporte más eficiente, pues reduce la electroósmosis y la adsorción. Se usa principalmente en la separación de proteínas y permite una separación más precisa según el tamaño molecular, similar a la cromatografía en gel. Las proteínas separadas se revelan con azul Coomassie 250.
Un soporte ideal debe ser:
- Químicamente inerte, sin interacción con la muestra ni con la solución tampón.
- No ionizable.
- Consistente y homogéneo.
- Estable y de bajo costo.
La electroforesis es una técnica de laboratorio que permite separar biomoléculas aplicando una corriente eléctrica a través de una matriz gelatinosa.
Métodos de detección
Una vez separadas las fracciones de la muestra, se utilizan distintos métodos para su detección y cuantificación:
- Fluorescencia o absorción UV-VISSe basa en el mismo principio que la espectroscopia de absorción y puede aplicarse directamente sobre las fracciones separadas o tras su tinción. La tinción puede realizarse antes o después del proceso electroforético.Una vez teñidas, las bandas obtenidas se analizan mediante densitometría, generando una representación gráfica de la posición y absorbancia de cada banda.
- Métodos radioquímicosLa muestra se marca previamente con un isótopo radiactivo. Tras la electroforesis, la detección se realiza midiendo la emisión de radiactividad.
- Métodos biológicosExisten dos enfoques principales:
- Detección de actividad enzimática.
- Detección de características antigénicas: se basa en la reacción antígeno-anticuerpo, que genera un precipitado visible (inmunoelectroforesis).
Fundamento
Cuando una mezcla de moléculas con carga eléctrica neta se coloca en un campo eléctrico, estas experimentan una atracción hacia el polo de carga opuesta. Con el paso del tiempo, las moléculas con carga positiva se moverán hacia el cátodo (polo negativo) y las de carga negativa hacia el ánodo (polo positivo).
El desplazamiento de las moléculas no solo está influenciado por el campo eléctrico, sino también por otras dos fuerzas: la fricción con el solvente, que se opone al movimiento, y el movimiento aleatorio de las moléculas (movimiento browniano), causado por su energía cinética. A medida que la temperatura aumenta, la energía cinética también lo hace, incrementando la difusión molecular.
Estas fuerzas combinadas hacen que las moléculas no se desplacen de manera uniforme, sino que formen un frente de migración cuya anchura aumenta con el tiempo. Para reducir este efecto, se emplean medios que generan mayor resistencia al movimiento molecular, como los geles. Un gel está compuesto por polímeros de alto peso molecular que retienen agua y crean una estructura porosa que limita la movilidad de los solutos. Esto disminuye la velocidad de migración, pero también evita que el frente se expanda demasiado.
Métodos Electroforéticos Zonales
Estos métodos son ampliamente utilizados debido a su capacidad para separar mezclas complejas. Se aplican pequeñas cantidades de solución de proteínas sobre un soporte sólido impregnado con una solución tampón. Los soportes suelen ser polímeros que forman un gel poroso, limitando el movimiento molecular y minimizando los flujos de convección del solvente.
Entre los materiales utilizados como soporte están el papel (celulosa), el almidón, la poliacrilamida, la agarosa y el acetato de celulosa. Este método es altamente eficiente, ya que se coloca una pequeña cantidad de muestra en una zona reducida, mientras que la distancia de migración es considerablemente mayor.
El equipamiento necesario incluye una fuente de alimentación, una cubeta de electroforesis (vertical u horizontal), un soporte y dos electrodos.
Electroforesis en Gel de Poliacrilamida
Los geles de poliacrilamida se obtienen mediante la polimerización de la acrilamida en presencia de un agente entrecruzador. Son químicamente inertes, presentan propiedades homogéneas y pueden prepararse de manera rápida y reproducible.
Estos geles son transparentes, mecánicamente estables, insolubles en agua y relativamente no iónicos, lo que permite una visualización clara de las bandas obtenidas. Una ventaja importante es que modificando la concentración de los polímeros se puede ajustar el tamaño de los poros, optimizando la separación de moléculas según sus características. Sin embargo, su uso en diagnóstico ha disminuido debido a su toxicidad neurológica.
Electroforesis en Geles de Gradiente
Se emplean geles de poliacrilamida con un gradiente progresivo de concentración de acrilamida y bisacrilamida, lo que genera una reducción gradual del tamaño del poro a lo largo del gel.
Las proteínas migran hasta alcanzar una zona donde el tamaño del poro impide su avance significativo. Aunque la migración no se detiene por completo, se reduce considerablemente.
Este tipo de geles mejora la resolución de las bandas, ya que las concentra en áreas más reducidas, y permite analizar un rango más amplio de pesos moleculares en una misma corrida electroforética, en comparación con los geles de concentración uniforme.
Electroforesis en Geles de Agarosa
La agarosa es un polisacárido de origen natural (derivado de algas como el agar-agar), cuya composición es homogénea. Sus disoluciones, generalmente con concentraciones entre 0,5 % y 2 %, permanecen líquidas por encima de los 50 °C y se solidifican al enfriarse, formando un gel semisólido.
Este gel está compuesto por una matriz tridimensional de fibras poliméricas embebidas en líquido, lo que ralentiza el movimiento de las moléculas. Se emplea principalmente para la separación de moléculas grandes, como ácidos nucleicos de aproximadamente 20.000 nucleótidos de longitud.
Electroforesis Capilar
La electroforesis capilar se basa en los mismos principios que las técnicas electroforéticas tradicionales, pero utiliza tecnología y condiciones diferentes que aportan varias ventajas. En este método, la separación de péptidos se realiza en un capilar de sílica fundida sometido a altos voltajes, generalmente entre 20 y 30 kV, en un campo de 400 a 500 V/cm, y con refrigeración.
La corriente electroendosmótica (FEO), generada por los grupos silanol de la superficie interna del capilar, origina un flujo uniforme de líquido, lo que contrasta con el flujo parabólico característico de la cromatografía líquida de alta resolución.
Una de las principales ventajas de esta técnica es que el capilar de sílica fundida, generalmente recubierto con una capa de polimina para incrementar su rigidez y resistencia, permite el paso de la luz ultravioleta, lo que facilita la visualización en tiempo real.
Este método posibilita la separación simultánea de cationes, aniones, proteínas, macromoléculas y sustancias sin carga.
Isoelectroenfoque
También conocido como electroenfoque, este método permite la separación de moléculas en un gradiente de pH. Las moléculas anfotéricas, como los aminoácidos, migran en un medio con una diferencia de potencial y un gradiente de pH. La zona del ánodo es ácida, mientras que la del cátodo es alcalina, estableciendo así un gradiente de pH en el que cada molécula se posiciona en el punto donde su carga neta es nula (punto isoeléctrico).
Las sustancias situadas en regiones de pH inferior a su punto isoeléctrico tienen carga positiva y migran hacia el cátodo, mientras que aquellas en zonas con pH superior a su punto isoeléctrico tienen carga negativa y se desplazan hacia el ánodo.
Cuando las moléculas alcanzan el área donde el pH coincide con su punto isoeléctrico, su carga neta se vuelve nula y su movimiento cesa. De esta forma, las moléculas anfotéricas se agrupan en bandas estrechas en función de su punto isoeléctrico.
Electroforesis para el Estudio de Ácidos Nucleicos
Electroforesis de Ácidos Nucleicos
El esqueleto de azúcar-fosfato de los ácidos nucleicos está cargado negativamente, lo que permite que el ADN o el ARN se desplace en respuesta a un campo eléctrico en función de su tamaño o longitud molecular. Este principio se usa para determinar el tamaño de los fragmentos de ácidos nucleicos mediante separación electroforética.
El procedimiento es similar al usado en la separación de proteínas, pero con algunas adaptaciones debido al mayor tamaño de las moléculas de ácidos nucleicos. El soporte de acrilamida suele ser insuficiente para estas moléculas, y además presentan diversas conformaciones estructurales.
Las muestras se cargan en pocillos ubicados en un extremo de un medio de separación sólido y poroso. Al aplicar el voltaje, los fragmentos se desplazan hacia el electrodo positivo de forma lineal, formando bandas migratorias. El medio en el que se produce la migración se conoce como gel, y los pocillos donde se colocan las muestras están dispuestos en serie.
Para facilitar la identificación de los fragmentos de ADN o ARN, se utilizan estándares de tamaño conocidos, denominados “escaleras de ADN o ARN”. Estas consisten en mezclas de ácidos nucleicos de longitud predefinida que sirven como referencia en la comparación de las muestras analizadas. La determinación del tamaño de los fragmentos se realiza comparando su migración con la de la escalera, ya sea mediante mediciones manuales o con el apoyo de software especializado.
El medio de separación y su concentración influyen en la resolución de los fragmentos en bandas diferenciadas. Otros factores que afectan la calidad de la separación incluyen el grosor del gel, la longitud de la carrera electroforética, la duración del proceso y el voltaje aplicado.
El gel de agarosa es el más utilizado para la electroforesis de ácidos nucleicos. Se coloca en una cubeta de electroforesis horizontal y se sumerge en una solución tampón. Las muestras se preparan con agentes espesantes como sacarosa o Ficoll antes de ser depositadas en los pocillos del gel.
La visualización de las bandas separadas se realiza mediante la tinción con colorantes intercalantes como el bromuro de etidio, que se inserta entre las bases emparejadas del ADN y se observa bajo luz ultravioleta utilizando un transiluminador.
Para que una banda sea visible, debe alcanzar una concentración suficientemente alta. En algunas aplicaciones, la detección de fragmentos de ácidos nucleicos se realiza mediante fluorescencia o radioactividad.
Las moléculas de ADN genómico pueden fragmentarse mediante digestión enzimática con enzimas de restricción, lo que permite su análisis por electroforesis. Las enzimas de restricción con sitios de reconocimiento simples generan fragmentos de menos de 50 kilopares de bases (Kbp), mientras que aquellas con secuencias más complejas pueden producir fragmentos de mayor tamaño (medidos en megapares de bases, Mpb). Para separar estos fragmentos de gran tamaño se utilizan sistemas electroforéticos con campos eléctricos pulsados.
En la mayoría de los análisis de ADN, se emplean condiciones no desnaturalizantes, lo que significa que los fragmentos permanecen en su conformación de doble hebra. En contraste, las separaciones de ARN suelen realizarse en condiciones desnaturalizantes para eliminar la estructura secundaria de las moléculas monocatenarias.
Secuenciación de ADN
El ADN es una molécula de doble hélice compuesta por dos cadenas formadas por millones de nucleótidos. Cada nucleótido está constituido por un azúcar (desoxirribosa), una base nitrogenada (A, T, G, C) y un grupo fosfato. La disposición específica de los nucleótidos dentro del ADN se denomina secuencia, y las técnicas utilizadas para determinar esta secuencia conforman el proceso de secuenciación.
Desde que Watson descubrió la estructura del ADN en 1953, los avances científicos y tecnológicos han permitido la obtención de numerosas secuencias de ADN con aplicaciones en diversos campos, como la medicina, la ecología, la fisiología y la evolución. En 1976, Walter Fiers y su equipo lograron secuenciar el genoma completo del bacteriófago MS2, compuesto por aproximadamente 3.500 pares de bases. Posteriormente, en 1977, Maxam y Gilbert revolucionaron la biología molecular con el desarrollo de la secuenciación enzimática de Sanger.
La técnica enzimática se basa en la interrupción controlada de la replicación del ADN en un entorno in vitro. Para ello, se realiza una amplificación por PCR del fragmento que se desea secuenciar. A diferencia de una PCR convencional, en esta técnica se emplea un solo cebador y se añaden dideoxinucleótidos (ddNTPs), los cuales carecen del grupo hidroxilo en el extremo 3′ y están marcados con radiactividad o fluoróforos. Cuando un ddNTP se incorpora a la cadena en crecimiento, la elongación se detiene, generando fragmentos de ADN de diferentes longitudes, cada uno finalizando con un ddNTP. Estos fragmentos se separan mediante electroforesis y se analizan de forma manual o automatizada.
El resultado de este proceso se representa en un electroferograma, un gráfico generado por el secuenciador tras el análisis electroforético. En este gráfico se muestra la disposición de las bases en función de curvas de fluorescencia específicas para cada base. Es fundamental revisar los electroferogramas antes de continuar con otros análisis, ya que proporcionan información sobre la calidad de la PCR y la confiabilidad de los datos obtenidos.
Una vez obtenida la secuencia de interés, esta se compara con secuencias almacenadas en bases de datos en línea para su análisis e interpretación.
Nota Importante
Para la separación de los fragmentos de ADN, el gel de agarosa se disuelve en un tampón TEB en el microondas. Una vez que la mezcla se vuelve transparente, se agrega un colorante como el bromuro de etidio para su posterior visualización.
3.5. Cromatografía
Las técnicas cromatográficas comprenden un conjunto de métodos empleados para separar, aislar e identificar los componentes de mezclas complejas, permitiendo también su cuantificación. Por esta razón, es una técnica ampliamente utilizada en distintos campos.
Las separaciones cromatográficas se basan en la adsorción selectiva de los distintos componentes de una mezcla en diferentes adsorbentes. El proceso se fundamenta en el transporte de la mezcla a través de una fase estacionaria (que puede ser sólida o líquida) mediante una fase móvil (que puede ser líquida o gaseosa), la cual fluye sobre la fase estacionaria. Así, la cromatografía se compone de dos fases principales: la fase estacionaria y la fase móvil.
La fase estacionaria es una sustancia con una gran superficie disponible, considerada en química como un gel. Su estructura esponjosa permite que las sustancias disueltas penetren en su interior y sean adsorbidas. Por otro lado, la fase móvil es la que se desplaza sobre la fase estacionaria, arrastrando los componentes de la muestra a distintas velocidades según sus características.
Existen diversas formas de realizar un proceso cromatográfico, dependiendo de la manera en que se introduce la muestra y su desplazamiento en la fase estacionaria. Los métodos más comunes son:
a) Elución.
b) Desplazamiento.
c) Análisis frontal.
El método de elución es el más utilizado y el que se explicará aquí. En este procedimiento, la muestra se deposita en la parte superior de una columna y la fase móvil fluye a través de ella, arrastrando los componentes de la muestra a diferentes velocidades. Los componentes menos retenidos por la fase estacionaria saldrán primero, mientras que los más retenidos lo harán después.
Clasificación
- Según la naturaleza de la fase móvil (FM):
- FM líquida: cromatografía líquida.
- FM gaseosa: cromatografía de gases.
- Según la naturaleza de la fase estacionaria (FE):
- FE sólida: cromatografía de adsorción.
- FE líquida: cromatografía de partición o reparto.
Existen numerosas técnicas cromatográficas (cromatografía en papel, en capa fina, de gases, en columna, etc.), pero todas pueden clasificarse según el tipo de interacción entre el soluto y la fase estacionaria. Así, se distinguen los siguientes tipos:
- Cromatografía de reparto.
- Cromatografía de adsorción.
- Cromatografía de intercambio iónico.
- Exclusión molecular.
Cromatografía de Reparto
En esta técnica, los solutos se separan en función de su distribución diferencial entre dos fases líquidas (cromatografía líquido-líquido) o entre un gas y una fase líquida (cromatografía gas-líquido). La fase estacionaria siempre es un líquido soportado en un sólido, mientras que la fase móvil puede ser un líquido o un gas. Un caso particular de cromatografía líquido-líquido es la cromatografía en papel, donde la fase estacionaria es una capa de agua fijada a una hoja de papel.
El procedimiento se lleva a cabo en tiras de papel de filtro o en capas delgadas de celulosa, gel de sílice húmedo o tierra de diatomeas. Estos materiales porosos sirven de soporte para la fase estacionaria, mientras que la fase móvil suele ser un disolvente orgánico no miscible con el agua. La solubilidad del compuesto en ambas fases influye en su movilidad durante el proceso.
Los principales tipos de cromatografía de reparto son:
a) Cromatografía en papel.
b) Cromatografía de gases.
c) Cromatografía líquido-líquido.
A) Cromatografía en Papel
Esta técnica pertenece al grupo de cromatografías de reparto líquido-líquido, aunque también pueden intervenir fenómenos de adsorción. El papel (celulosa) actúa como fase estacionaria al retener agua en sus fibras. La fase móvil suele ser menos polar y está compuesta por disolventes orgánicos y acuosos. Actualmente, su uso es limitado.
Es un método que requiere pequeñas cantidades de muestra y se emplea para la identificación cualitativa de sus componentes.
El procedimiento consiste en depositar una pequeña cantidad de la muestra en el extremo de una tira de papel y dejar que el disolvente la atraviese en un espacio cerrado saturado con su vapor. La velocidad de desplazamiento de cada componente dependerá de su solubilidad en la fase móvil y de su interacción con la fase estacionaria. Una vez que el disolvente ha recorrido la distancia requerida, la tira se seca y se analiza.
Si los componentes son coloreados, la separación es fácilmente visible. En caso contrario, se emplean métodos químicos para su revelado. Para cuantificar el desplazamiento, se usa el factor de retardación (Rf), que se calcula con la siguiente fórmula:
Rf = Distancia recorrida por la sustancia / Distancia recorrida por el disolvente
Para mejorar la separación de los componentes, se puede realizar una cromatografía bidimensional, en la que la muestra se somete a dos procesos sucesivos en direcciones perpendiculares.
Aplicaciones
- Separación de aminoácidos.
- Resolución de problemas en química orgánica, inorgánica y bioquímica.
- Identificación de metales en muestras minerales.
- Control de pureza en productos farmacéuticos y alimenticios.
B) Cromatografía de Gases
Esta técnica es una de las más utilizadas y permite la separación de compuestos gaseosos, líquidos volátiles y sólidos en estado de vapor. En este proceso, la muestra debe encontrarse en estado gaseoso, y la fase móvil es un gas inerte que actúa como portador.
Tipos de Cromatografía de Gases
- Cromatografía sólido-gas (CSG): la fase estacionaria es un sólido. Su uso es limitado.
- Cromatografía líquido-gas (CLG):
- Fase estacionaria: un líquido no volátil impregnado en un soporte inerte dentro de la columna.
- Muestra: debe ser volátil.
- Fase móvil: un gas inerte (como nitrógeno o argón), cuyo flujo y presión deben controlarse para optimizar el rendimiento de la columna.
Este método se emplea en numerosas aplicaciones, incluyendo la identificación de compuestos en mezclas complejas y el análisis cuantitativo de sustancias volátiles.
Control de la Temperatura en Cromatografía de Gases
El control preciso de la temperatura es un factor clave en la cromatografía de gases, ya que influye directamente en la separación de los componentes de una mezcla.
Los compuestos de la muestra se inyectan en un flujo de gas inerte (como nitrógeno, hidrógeno, argón o helio), que los transporta a través de una columna especializada. Esta columna está rellena con un material poroso recubierto por una fina película de una fase líquida no volátil, la cual actúa como fase estacionaria. Entre las sustancias más utilizadas para esta fase estacionaria se encuentran los aceites de silicona y aceites parafínicos.
A medida que los componentes atraviesan la columna, se separan debido a las diferencias en su interacción con la fase estacionaria. Una vez separados, emergen de la columna en diferentes momentos y son detectados individualmente. La identificación de los componentes se realiza midiendo el tiempo de retención, es decir, el tiempo transcurrido desde la inyección de la muestra hasta la aparición del pico en el registrador. Comparando estos tiempos con los de sustancias puras de referencia, se puede identificar cada compuesto. Además, el área bajo el pico en el cromatograma es proporcional a la concentración del componente.
Componentes del Sistema de Cromatografía de Gases
El sistema cromatográfico se compone principalmente de:
- Columnas: Pueden tener forma de U, W o espiral, permitiendo su colocación eficiente dentro del horno de calentamiento. Estas columnas están fabricadas en diversos materiales, como plástico (poco resistente), metal (las más utilizadas) o vidrio (económicas, inertes, pero frágiles).
- Detectores: Existen más de 25 tipos de detectores, algunos con gran sensibilidad. Se dividen en:
- Diferenciales: Miden la concentración del componente en el momento de su salida.
- Integrales: Acumulan los componentes eluidos hasta un punto determinado.
- Destructivos y no destructivos: Según su impacto sobre la muestra analizada.
Aplicaciones de la Cromatografía de Gases
Esta técnica se emplea en el análisis de moléculas orgánicas en sus formas neutras, así como en el estudio de ácidos y bases libres (por ejemplo, fármacos y ácidos orgánicos en plasma). Es fundamental que la muestra posea suficiente volatilidad para ser analizada mediante este método.
Cromatografía Líquida en Columna
Conceptos Claves en Cromatografía
- Tiempo de retención (tr): Intervalo de tiempo desde la inyección de la muestra hasta su salida de la columna.
- Volumen de elución (Ve): Cantidad de fase móvil requerida para que un soluto eluya.
- Volumen de exclusión (Vo): Volumen de elución de un soluto no retenido.
- Anchura del pico (W): Amplitud del pico en el cromatograma.
- Resolución (R): Nivel de separación entre dos picos consecutivos.
Fórmula de la Resolución:
Una resolución de 1,25 se considera aceptable para un análisis confiable.
Eficiencia de la columna:
La eficiencia de una columna se evalúa en función de la nitidez de los picos obtenidos. Se mide utilizando el concepto de platos teóricos (N), los cuales representan zonas de equilibrio entre el soluto y las fases estacionaria y móvil.
Altura Equivalente a un Plato Teórico (AEPT):
Donde L es la longitud de la columna. Un mayor número de platos teóricos y una menor AEPT indican una mayor eficiencia de la columna.
Una vez que el componente atraviesa la columna, eluye en un tiempo determinado y genera un pico en el cromatograma. La detección puede realizarse mediante métodos electroquímicos, calorimétricos, fluorimétricos, entre otros.
Tipos de Cromatografía Líquida
- Cromatografía líquida clásica: La fase móvil fluye por gravedad a través de la columna.
- HPLC (Cromatografía Líquida de Alta Resolución): Se emplea una bomba para aumentar la presión y mejorar el flujo de la fase móvil. Un esquema de la instrumentación utilizada en HPLC se muestra en la figura.
- Cromatografía líquida isocrática: Se utiliza una fase móvil de composición constante durante todo el análisis.
- Cromatografía líquida en gradiente de elución: La composición de la fase móvil varía progresivamente a lo largo del proceso cromatográfico.
Aplicaciones de la Cromatografía Líquida
- Análisis Cuantitativo: Se mide la altura o el área del pico y se compara con una muestra patrón.
- Análisis Cualitativo: Se evalúa la posición del pico en el cromatograma, ya que el tiempo de retención es característico para cada sustancia.
Cromatografía de Adsorción
La separación de los componentes de una mezcla se basa en las diferencias en sus interacciones con la fase estacionaria, que es un sólido en cuya superficie ocurren interacciones específicas con las moléculas de la fase móvil (líquido o gas). Estas interacciones pueden incluir enlaces de hidrógeno o fuerzas electrostáticas. Este tipo de interacciones también se observa en técnicas de cromatografía planar.
Cromatografía en Capa Fina
En esta técnica, en lugar de papel, se utiliza un material inerte como sílice gel o gel de óxido de aluminio, que se extiende sobre una base de cristal o vidrio en forma de una película delgada.
Las interacciones entre la fase móvil y la fase estacionaria pueden implicar fenómenos de adsorción (si la placa está seca) o una combinación de adsorción y partición (si el material está húmedo). La muestra se deposita en un extremo de la placa, que luego se coloca en un recipiente con el líquido de desarrollo (fase móvil).
Las sustancias con mayor solubilidad en la fase estacionaria quedan más retenidas que aquellas que son más solubles en la fase móvil. Esto genera un desplazamiento característico y reproducible de los compuestos, medido mediante el factor de retardo (Rf o Rx).
El desarrollo cromatográfico puede ser:
- Monodimensional o bidimensional
- Ascendente, descendente o horizontal, según la dirección del flujo de la fase móvil.
Una vez completado el proceso, los compuestos se identifican visualmente si presentan color o fluorescencia, o mediante un revelado con reactivos específicos (por ejemplo, vapores de yodo o ninhidrina). El análisis cualitativo permite identificar la presencia de un compuesto, mientras que el análisis cuantitativo o semicuantitativo compara la concentración del compuesto con un estándar de referencia.
Aplicaciones
- Fraccionamiento de lípidos (por ejemplo, fosfolípidos en líquido amniótico).
- Separación de aminoácidos.
Cromatografía de Intercambio Iónico
En este método, la fase estacionaria interactúa con los componentes de la mezcla mediante atracciones iónicas entre cargas opuestas. Los intercambiadores iónicos más comunes son resinas poliméricas sintéticas, generalmente en forma de esferas de diferentes tamaños. Estas resinas pueden modificarse con grupos químicos de carga positiva o negativa para crear un lecho de resina con propiedades polianiónicas o policatiónicas.
Los iones de la muestra compiten con los de la fase móvil por los sitios de unión en la fase estacionaria. Los iones con mayor afinidad se retienen más tiempo y eluyen más tarde, mientras que los de menor interacción salen primero de la columna. El grado de ionización de los compuestos se controla ajustando el pH de la fase móvil.
Tipos de Cromatografía de Intercambio Iónico
A) Cromatografía de Intercambio Catiónico
En este tipo de cromatografía, la fase estacionaria contiene grupos ácidos (por ejemplo, ácido sulfónico -SO₃H) que intercambian cationes presentes en la muestra.
Mecanismo de Intercambio:
Res – SO₃H⁺ + A⁺ → Res – SO₃A⁺ + H⁺
B) Cromatografía de Intercambio Aniónico
Aquí, la fase estacionaria tiene grupos básicos con carga positiva (como sales de amonio cuaternario), que intercambian aniones presentes en la muestra.
Mecanismo de Intercambio:
Res – CH2N⁺(CH₃)₃Cl⁻ + A⁻ → Res – CH2+N(CH₃)₃A⁻ + Cl⁻
Aplicaciones
- Purificación de agua con iones disueltos (ablandamiento y desionización del agua).
- Separación de aminoácidos de otras especies iónicas.
- Eliminación de sustancias interferentes antes de realizar otros análisis.
Cromatografía de Exclusión o Tamizado Molecular
También conocida como filtración en gel, esta técnica separa los compuestos según su peso molecular. La fase estacionaria consiste en geles poliméricos con una estructura tridimensional que contiene poros de un tamaño específico.
Las moléculas de mayor tamaño, que no pueden penetrar en los poros, eluyen rápidamente sin ser retenidas en la fase estacionaria. En cambio, las moléculas más pequeñas entran en los poros, lo que prolonga su recorrido y retrasa su elución.
La elección del tipo de gel depende del peso molecular de la muestra y su compatibilidad con la fase móvil.
Aplicaciones
- Separación de proteínas marcadas.
- Determinación de pesos moleculares.
- Eliminación de sustancias interferentes en muestras biológicas y químicas.
Calibración y mantenimiento
La calibración en los sistemas automatizados de laboratorio es fundamental para garantizar resultados precisos y confiables. Desde el momento en que se instala un instrumento analítico, sin importar la técnica que emplee, es necesario realizar de manera programada al menos las siguientes acciones:
- Calibración
- Verificación
- Mantenimiento
- Control
La calibración se define como el conjunto de procedimientos aplicados a un equipo de medición con el propósito de asegurar que cumple con las especificaciones de precisión requeridas. Este proceso implica comparar la respuesta del instrumento con un material de referencia de propiedades conocidas y, si es necesario, aplicar un factor de corrección para obtener un valor adecuado.
El resultado de la calibración permite identificar posibles errores en la medición del instrumento, del sistema de medición o de la propia muestra analizada. Debido a que las mediciones realizadas en estos equipos pueden presentar ciertas imprecisiones, lo que incrementa la incertidumbre en los análisis, es esencial contar con un Plan de Calibración para minimizar estos errores.
Plan de Calibración
El laboratorio debe determinar qué equipos de medición y ensayo requieren calibración debido a su influencia en los resultados. Para ello, se establece y mantiene un Plan de Calibración, en el que se detallan las actividades a realizar y su frecuencia.
Procedimiento de calibración
La calibración debe realizarse siguiendo instrucciones documentadas. En algunos casos, estos procedimientos están regulados por organismos nacionales e internacionales, mientras que en otros, las directrices son proporcionadas por los fabricantes. Cada protocolo de calibración debe incluir los siguientes aspectos:
- Objetivo
- Generalidades y alcance
- Materiales necesarios
- Requisitos previos y recomendaciones
- Procedimiento a seguir
- Registro de datos obtenidos
- Cálculo de resultados e incertidumbres
- Evaluación de los resultados
- Certificado de calibración
- Identificación del estado de calibración
De manera periódica, se realizan mediciones en los equipos con materiales de referencia adecuados. El laboratorio debe registrar los resultados de cada calibración en un documento que contenga la información detallada del procedimiento realizado, incluyendo la identificación del equipo calibrado, el método empleado, los patrones de calibración utilizados, las condiciones ambientales, los resultados obtenidos, la incertidumbre asociada y la persona responsable del proceso.
Gases para calibración en cromatografía de gases
En cromatografía de gases, la precisión depende tanto de la calidad de los gases utilizados como del desempeño del equipo.
Las impurezas en el flujo de gas pueden generar problemas como contaminación en las columnas y detectores, inestabilidad en la línea base, aparición de picos no deseados y errores en los resultados. Estas impurezas pueden deberse a una selección inadecuada del grado de pureza del gas, al uso de materiales incorrectos en el sistema de suministro o a la presencia de fugas.
El grado de pureza necesario dependerá de la aplicación, ya que, a mayor sensibilidad y selectividad, mayor será la exigencia en la pureza del gas. Entre los problemas más comunes que afectan el rendimiento del equipo se encuentran:
- Deterioro de la columna: Puede provocar una resolución deficiente, inestabilidad en la línea base, desplazamiento de picos y acumulación de residuos. Los principales responsables de estos problemas suelen ser el oxígeno y la humedad presentes en el gas portador.
- Contaminación orgánica: Sustancias como los hidrocarburos pesados pueden generar picos fantasmas, alterando los resultados analíticos.
- Desempeño del detector: Impurezas en el gas portador o en los gases auxiliares pueden afectar la sensibilidad del detector, generar ruido en la señal, provocar desviaciones y reducir su vida útil.
Efectos de las impurezas en los detectores
Cada tipo de detector empleado en cromatografía de gases puede verse afectado de diferentes maneras por la presencia de impurezas en el gas utilizado:
- Detector de conductividad térmica (TCD): El oxígeno puede oxidar el filamento, reduciendo la sensibilidad y acortando su vida útil.
- Detector de ionización de llama (FID): Los hidrocarburos pueden aumentar el ruido y la corriente de fondo, disminuyendo la sensibilidad y provocando desviaciones en la línea base o la aparición de picos no deseados.
- Detector de captura de electrones (ECD): La presencia de compuestos halogenados, oxígeno o agua puede incrementar el ruido y las corrientes de fondo, reduciendo la sensibilidad y afectando la estabilidad de la columna.
- Detector de fotoionización (PID): Impurezas con carga negativa pueden disminuir la señal del detector, mientras que los hidrocarburos pueden ensuciar la ventana óptica.
- Detector de ionización por helio (HID): La presencia de impurezas reduce la sensibilidad del detector y puede generar picos negativos, especialmente si se trata de oxígeno o humedad.
- Detector de descarga de ionización (DID): La contaminación por gases permanentes puede disminuir significativamente la sensibilidad de este detector.
- Detector fotométrico de llama (FPD): Los hidrocarburos pueden alterar la respuesta del detector, afectando la precisión de la medición.
Garantía de pureza mediante el uso de equipos adecuados
Para mantener la pureza del gas, es fundamental utilizar un equipo adecuado para su manejo. Algunos materiales pueden liberar impurezas o permitir su filtración en el flujo de gas, afectando los resultados del análisis.
Reguladores
Los reguladores de gas deben contar con filtros en la entrada para evitar la entrada de partículas contaminantes, las cuales pueden quedar atrapadas y provocar un aumento en la presión de salida.
El diafragma del regulador debe estar fabricado en acero inoxidable en lugar de materiales como el neopreno, que es permeable al oxígeno y la humedad y puede liberar compuestos orgánicos no deseados.
Válvulas
Los materiales utilizados en la fabricación de válvulas deben cumplir con los mismos criterios que los empleados en los reguladores.
Es importante considerar el diseño de la válvula, ya que pueden ser de dos tipos:
- Válvulas con empaque: Utilizan un material de sellado que impide la fuga de gas, pero con el uso prolongado pueden presentar pequeñas filtraciones que permiten la entrada de aire y humedad, contaminando el sistema.
- Válvulas sin empaque: Incorporan un diafragma o un sello metálico de fuelle, lo que evita la contaminación atmosférica en el flujo del gas.
3.6. Inmunoensayos
Métodos basados en la interacción antígeno-anticuerpo
Dentro de las técnicas inmunológicas, las más relevantes son aquellas que dependen de la unión específica entre antígeno (Ag) y anticuerpo (Ac). Las inmunoglobulinas tienen la capacidad de reconocer y unirse a un antígeno de manera específica, lo que permite detectar dicha unión mediante diversos métodos. Entre ellos, se incluyen la precipitación, la aglutinación y otros procedimientos indirectos como la fluorescencia, el uso de radioisótopos o la aplicación de enzimas.
Los antígenos y anticuerpos se caracterizan por su interacción mutua, lo que permite emplear uno de ellos para la detección o cuantificación del otro. Existen múltiples técnicas que emplean distintos enfoques para visualizar y medir esta unión, lo que las convierte en herramientas esenciales en inmunodiagnóstico y análisis biológico.
Técnicas de aglutinación: directa, indirecta, inhibición y Coombs
Las técnicas de aglutinación permiten visualizar los complejos formados por la unión entre antígenos (Ag) y anticuerpos (Ac), ya que los Ac provocan la aglutinación cuando los Ag están presentes en la superficie de glóbulos rojos, plaquetas, leucocitos o partículas inertes como el látex. Los métodos más utilizados emplean glóbulos rojos (hemoaglutinación) o partículas de látex. En estas reacciones, el anticuerpo específico se denomina aglutinina.
Tipos de aglutinación
- Aglutinación directa o activa: Se produce cuando los antígenos ya tienen una estructura particulada de forma natural. Ejemplos de estos antígenos son las bacterias y los hematíes.
- Aglutinación indirecta o pasiva: Ocurre cuando los antígenos deben ser fijados a la superficie de partículas inertes, como látex o bentonita, para generar la reacción de aglutinación.
- Anticuerpo completo: Es aquel que, al unirse a su antígeno específico, provoca la aglutinación.
- Anticuerpo incompleto: Se une a su antígeno específico, pero no desencadena la aglutinación.
Características de las reacciones de aglutinación
- Son técnicas con alta sensibilidad, permitiendo detectar pequeñas cantidades de Ag o Ac.
- Tienen menor especificidad en comparación con las reacciones de precipitación.
- Pueden ser utilizadas como pruebas cualitativas o semicuantitativas.
- Son procedimientos prácticos, ya que permiten fijar Ag o Ac a partículas para facilitar su detección.
- La aglutinación puede observarse a simple vista.
Factores que influyen en la aglutinación
- Carga de las partículas:
- Las partículas en suspensión tienen una carga superficial negativa (potencial zeta) que provoca su repulsión mutua.
- La unión con los Ac forma enlaces entre partículas, favoreciendo la aglutinación.
- En algunos casos, es necesario reducir el potencial zeta mediante:
- Aumento de la viscosidad con albúmina.
- Tratamiento con enzimas proteolíticas cuando las partículas son hematíes.
- Uso de buffers que disminuyan la carga negativa de las partículas.
- Tipo de anticuerpo:
- Las inmunoglobulinas IgM, debido a su tamaño, generan siempre aglutinación.
- Las IgG pueden o no producir aglutinación, dependiendo de la subclase y otras condiciones. Si no aglutinan, se denominan anticuerpos incompletos.
- Concentración de electrolitos:
- La fuerza iónica del medio afecta la carga superficial de las partículas.
- Para mejorar la aglutinación, se recomienda una fuerza iónica baja en el buffer.
- Se puede utilizar tripsina para disminuir la fuerza iónica del medio de reacción.
- pH:
- Debe mantenerse cercano al valor fisiológico para una reacción óptima.
- Viscosidad:
- Un medio más viscoso favorece la aglutinación.
- Se pueden agregar sustancias como albúmina o dextrano para aumentar la viscosidad.
- Antígenos:
- Un antígeno con un único determinante antigénico no puede inducir aglutinación.
- Cuantos más determinantes antigénicos tenga, mayor será la aglutinación.
- Temperatura y tiempo de incubación:
- Se recomienda una temperatura cercana a la fisiológica, aunque en algunos casos puede ser óptima a 4°C.
- Un tiempo de incubación prolongado favorece una mejor reacción.
- Agitación:
- Métodos que mejoran el contacto entre Ag y Ac, como la agitación o la centrifugación, facilitan la aglutinación.
Dato interesante
El potencial zeta es la carga negativa que presentan en su superficie los hematíes, bacterias y partículas inertes, lo que influye en la capacidad de aglutinación.
Tipos de reacciones de aglutinación
A) Aglutinación directa
Este tipo de aglutinación ocurre cuando un anticuerpo (Ac) se une a su antígeno (Ag) específico que ya está presente en forma particulada de manera natural. Puede emplearse tanto en análisis cualitativos como cuantitativos.
- a) Cualitativa:
Se lleva a cabo en un portaobjetos o tubo de ensayo, enfrentando un anticuerpo con una suspensión de antígenos, en la que uno de ellos es desconocido. Tras una breve agitación, se observa si ocurre aglutinación. Es una técnica comúnmente utilizada para identificar bacterias y determinar grupos sanguíneos. - b) Cuantitativa:
Permite estimar aproximadamente la cantidad de anticuerpos presentes en una muestra de suero, a través de la determinación del título de anticuerpos. Se usa con frecuencia en el diagnóstico serológico de infecciones como Brucella y Salmonella.
Es importante considerar el efecto zona, que ocurre cuando hay un exceso de anticuerpos en la muestra, lo que puede provocar la solubilización del aglutinado. Para evitar este problema, se recomienda utilizar una serie de tubos con diferentes diluciones.
B) Aglutinación indirecta
En esta técnica, los anticuerpos reaccionan con antígenos solubles que han sido fijados artificialmente a la superficie de partículas como eritrocitos (humanos o de origen animal), bacterias o partículas inertes (látex, bentonita, entre otras). La absorción del antígeno sobre la partícula convierte la reacción de precipitación Ag-Ac en una reacción de aglutinación, lo que mejora significativamente la sensibilidad del ensayo.
Se ha demostrado que las pruebas que emplean eritrocitos como soporte del antígeno son más sensibles que aquellas que utilizan partículas como látex o bentonita. Al igual que en la aglutinación directa, este método puede aplicarse en pruebas cualitativas o cuantitativas.
Existe una variante de esta técnica conocida como aglutinación indirecta inversa, en la que, en lugar de fijar el antígeno a la partícula, se acopla el anticuerpo específico del antígeno buscado.
Este procedimiento es ampliamente utilizado en serología debido a su rapidez, bajo costo y alta sensibilidad. Sin embargo, presenta el inconveniente de una menor especificidad, lo que puede generar falsos positivos.
Algunas de las pruebas más conocidas que emplean esta técnica son:
- Pruebas reumáticas, como la detección de proteína C reactiva, factores reumatoides (pruebas de Waaler-Rose y Singer-Plotz) y antiestreptolisina.
- RPR y VDRL, utilizadas en el diagnóstico de la sífilis.
- TPHA, empleada en la detección de la sífilis.
- Prueba de látex para Cryptococcus, utilizada en el diagnóstico de infecciones fúngicas.
Dato adicional
Las técnicas de aglutinación no requieren el marcado de moléculas específicas. La aglutinación se observa cuando los anticuerpos reaccionan con antígenos que forman parte de estructuras celulares como glóbulos rojos, plaquetas o leucocitos, o cuando han sido fijados artificialmente a estas estructuras.
C) Inhibición de la aglutinación
Los anticuerpos tienen la capacidad de reconocer y reaccionar con sus antígenos específicos. Sin embargo, cuando el antígeno se encuentra en una forma soluble dentro de la muestra, la unión entre Ag y Ac puede ocurrir sin que se observe aglutinación.
Este método es una variante de las reacciones de aglutinación y se emplea para detectar y cuantificar antígenos solubles.
Proceso de la prueba
La prueba se desarrolla en dos fases:
- Primera fase
- Se añade a la muestra problema el anticuerpo específico del antígeno que se busca detectar.
- Si no se observa aglutinación, pueden darse dos situaciones:
- El antígeno no está presente en la muestra.
- El antígeno está presente, pero en forma soluble.
- Segunda fase
- Se incorporan partículas recubiertas con el antígeno buscado. Dependiendo de la reacción observada, pueden ocurrir dos escenarios:
- Si se produce aglutinación: Indica que el antígeno no estaba presente en la muestra original, ya que los sitios de unión del anticuerpo estaban libres y reaccionaron con las partículas añadidas.
- Si no se produce aglutinación: Significa que el antígeno sí estaba presente en la muestra inicial en forma soluble. Como los sitios de unión del anticuerpo fueron ocupados por este antígeno, no pudieron interactuar con las partículas añadidas en la segunda etapa.
- Se incorporan partículas recubiertas con el antígeno buscado. Dependiendo de la reacción observada, pueden ocurrir dos escenarios:
Esta prueba también puede aplicarse de manera cuantitativa mediante la realización de diluciones sucesivas de la muestra problema. Un ejemplo práctico de este método es el diagnóstico del embarazo, en el que se detecta la presencia de la hormona GCh, que es un antígeno soluble.
D) Prueba de Coombs o detección de anticuerpos no aglutinantes
En algunos casos, ciertas inmunoglobulinas del tipo IgG pueden unirse a los antígenos de una partícula sin generar aglutinación. Estos anticuerpos son conocidos como anticuerpos incompletos.
La prueba de Coombs permite detectar estos anticuerpos mediante la adición de antiglobulinas IgG (también llamadas antiinmunoglobulinas), las cuales actúan como puentes para facilitar la aglutinación de las partículas.
Existen dos tipos de prueba:
a) Prueba de Coombs directa
Se utiliza para identificar anticuerpos que ya están adheridos a la superficie de células o partículas.
- Un ejemplo es la enfermedad hemolítica del recién nacido por incompatibilidad Rh. En estos casos, los glóbulos rojos del bebé han sido sensibilizados por anticuerpos maternos IgG, que no provocan aglutinación por sí solos.
- Al añadir antiglobulinas humanas, se establecen puentes entre los glóbulos rojos, permitiendo la aglutinación y confirmando la presencia de estos anticuerpos en la sangre del recién nacido.
b) Prueba de Coombs indirecta
Se utiliza para detectar anticuerpos no aglutinantes que circulan libres en el suero.
El procedimiento consta de dos etapas:
- Primera fase
- Se mezcla la muestra problema con antígenos particulados específicos.
- No se observa aglutinación en esta etapa.
- Segunda fase
- Se agregan antiglobulinas (anti-IgG).
- Estas se unen a los anticuerpos no aglutinantes que han quedado fijados a los antígenos, permitiendo que se produzca la aglutinación.
Este método es útil en el diagnóstico serológico de la brucelosis crónica y también se emplea para detectar anticuerpos maternos no aglutinantes en casos de incompatibilidad Rh.
Técnicas de precipitación
Las técnicas de precipitación se utilizan para detectar y cuantificar antígenos y anticuerpos mediante la formación de complejos insolubles en un medio fluido. Dentro de este grupo se incluyen:
- Métodos de precipitación propiamente dichos
- Técnica de inmunodifusión radial de Ouchterlony
- Método de inmunoelectroforesis
- Técnica de difusión radial simple de Mancini
Para llevar a cabo estas pruebas, es fundamental que tanto el antígeno (Ag) como el anticuerpo (Ac) se encuentren en una solución que permita la precipitación del complejo Ag-Ac.
Formación del precipitado
Cuando un antígeno y un anticuerpo específicos se combinan, pueden formar un complejo insoluble que precipita. Esto se explica mediante la teoría de la red, que establece que:
- Los antígenos poseen múltiples determinantes antigénicos.
- Los anticuerpos tienen varios sitios de unión.
- La interacción entre ellos genera una red molecular que, al aumentar su tamaño y complejidad, se vuelve insoluble y precipita.
La cantidad de precipitado dependerá de la relación entre las concentraciones de antígeno y anticuerpo.
- Exceso de anticuerpo (efecto prozona): Cada molécula de antígeno se une fácilmente a un anticuerpo, formando pocos enlaces cruzados y generando precipitados pequeños o inexistentes.
- Zona de equivalencia: Cuando las concentraciones de antígeno y anticuerpo son similares, se forman complejos de gran tamaño, lo que genera la máxima precipitación.
- Exceso de antígeno (efecto postzona): Cada anticuerpo puede fijar solo una molécula de antígeno libre, reduciendo los entrecruzamientos y la formación de precipitados.
Factores que afectan la precipitación
Algunas variables pueden influir en la reacción:
- Carga del complejo Ag-Ac
- Peso molecular del material proteico, siendo óptimo entre 40.000 y 160.000 dalton
- Temperatura de la reacción
- Medio de reacción, ya sea sólido o líquido
Técnica de inmunodifusión radial simple (Método de Mancini)
Este método es utilizado para cuantificar antígenos de manera precisa y se realiza en varias etapas:
- Preparación del medio:
- Se mezcla el anticuerpo específico con agar fundido.
- Se vierte la solución en placas de Petri o láminas de vidrio hasta solidificarse.
- Se perforan pequeños pocillos en el agar.
- Añadir muestras y estándares:
- Se colocan en los pocillos soluciones con antígeno de concentración conocida (estándares) y las muestras problema.
- El antígeno difunde a través del agar y, al encontrarse con el anticuerpo en la proporción adecuada, se forma un anillo de precipitado circular.
- Medición y análisis:
- El diámetro del anillo de precipitación es proporcional a la cantidad de antígeno presente.
- Se elabora una curva de referencia con los estándares, donde:
- En el eje X se representa el cuadrado del diámetro del anillo.
- En el eje Y se grafica la concentración del antígeno.
- A partir de esta curva, se determina la concentración de antígeno en la muestra problema.
Aplicaciones
Esta técnica es utilizada principalmente para la cuantificación de proteínas plasmáticas y otros componentes en estudios serológicos.
Dato clave
El método de Mancini es una técnica cuantitativa de inmunodifusión radial ampliamente utilizada para la determinación de antígenos en muestras biológicas.
Técnicas de Difusión Radial Doble de Ouchterlony
Este método se lleva a cabo en placas de agar, donde se realizan varios pocillos. En uno de ellos se deposita la muestra que se desea analizar, mientras que en los otros se coloca un anticuerpo específico frente a la sustancia a identificar.
Tanto el antígeno como el anticuerpo se colocan en pocillos separados dentro de la placa de agar. Durante la incubación, ambos componentes comienzan a difundirse por el medio. Cuando alcanzan una concentración equivalente, se forma un arco de precipitación, que indica la presencia del complejo Ag-Ac.
Una de las ventajas de esta técnica es que permite analizar la relación entre distintas mezclas antigénicas a través de la forma que adquieren las líneas de precipitación. Se pueden presentar tres escenarios:
- Reacción de identidad total
- Se da cuando el mismo antígeno está presente en ambos pocillos.
- Se forma una banda de precipitación en forma de un arco continuo.
- Reacción de identidad parcial
- Los antígenos analizados comparten una parte de su estructura (epítopo común), pero uno de ellos tiene una región adicional que también es reconocida por el suero.
- En este caso, el arco de precipitación presenta un espolón.
- Reacción de no identidad
- Cuando los antígenos en ambos pocillos son completamente diferentes, se generan bandas de precipitación independientes sin continuidad entre ellas.
Técnicas de Inmunoelectroforesis
Este procedimiento se emplea para detectar alteraciones cualitativas en proteínas presentes en distintos fluidos biológicos, como suero, orina o líquido cefalorraquídeo (LCR).
El proceso se lleva a cabo en dos etapas principales:
- Separación de los antígenos mediante electroforesis
- Se coloca la muestra en un soporte adecuado y se somete a un campo eléctrico.
- Los antígenos migran a través del medio según su carga y tamaño.
- Reacción con anticuerpos específicos
- En un pocillo adyacente, se deposita un antisuero dirigido contra una o varias proteínas a identificar.
- Los antígenos y anticuerpos difunden y, cuando se encuentran en concentraciones óptimas, forman arcos de precipitación característicos.
Los patrones de precipitación obtenidos se comparan con un estándar, permitiendo la identificación de los antígenos presentes en la muestra problema.
Este método es útil para detectar alteraciones cualitativas en proteínas específicas o analizar la composición proteica en líquidos biológicos.
Electroinmunodifusión Simple o Inmunoelectroforesis en Cohete (Rocket)
Este método es una variante de la inmunodifusión simple que emplea corriente eléctrica para mejorar la difusión de los antígenos y obtener resultados cuantificables.
Procedimiento
- Se prepara un medio gelificado que contenga anticuerpos.
- Se colocan pocillos en el gel, distribuyéndolos perpendicularmente al sentido del campo eléctrico.
- Se depositan en los pocillos las muestras problema y los estándares de referencia.
- Al aplicar corriente eléctrica, los antígenos migran a través del gel.
- A medida que se desplazan, reaccionan con los anticuerpos presentes en el gel, formando líneas de precipitación en forma de cono o cohete.
La distancia entre el pocillo y la punta del cono es directamente proporcional al logaritmo de la concentración del antígeno en la muestra analizada.
Este método es ampliamente utilizado para la cuantificación precisa de proteínas en distintas muestras biológicas.
Contrainmunoelectroforesis o doble electroinmunodifusión
En este método, la muestra no se deja difundir de manera espontánea, sino que se somete a un proceso de electroforesis, lo que acelera considerablemente la prueba. Para ello, el pocillo que contiene el anticuerpo se coloca en el polo positivo, mientras que el pocillo con el antígeno se ubica en el polo negativo. Durante la migración de ambas sustancias hacia los polos opuestos, se produce la formación del precipitado, que indica la reacción entre el antígeno y el anticuerpo.
Inmunoelectroforesis bidimensional
Esta técnica se emplea principalmente en investigación para la separación analítica de antígenos con características electroforéticas similares, como variantes genéticas de proteínas o moléculas implicadas en la coagulación, como el factor VIII.
El procedimiento consta de dos etapas, ambas basadas en la electroforesis:
- Separación de los antígenos
- La muestra, que puede ser suero, orina o líquido cefalorraquídeo, se somete a electroforesis en un gel de agarosa para separar sus componentes según su carga.
- Interacción con anticuerpos
- La tira de agarosa con las proteínas separadas se transfiere a una placa más grande.
- Se añade agarosa líquida con anticuerpos específicos para los antígenos que se desean analizar.
- Al difundirse, se forman arcos de precipitación que permiten identificar las proteínas presentes en la muestra.
Técnicas de precipitación en medio líquido
Prueba del anillo
En este ensayo, cuando se mezclan un antígeno soluble y su anticuerpo específico, se genera un precipitado visible si ambos se encuentran en la proporción adecuada.
Este método puede aplicarse de forma cualitativa o cuantitativa:
- Cualitativa
- Se coloca el anticuerpo en el fondo de un tubo de ensayo.
- Se añade el antígeno en la parte superior.
- Si el antígeno está presente en la muestra, se forma un disco blanquecino en la zona de contacto entre ambas sustancias.
- Se utiliza para detectar la proteína C reactiva o para la identificación de los grupos de Lancefield en estreptococos.
- Cuantitativa
- Se emplea el mismo procedimiento, pero utilizando diferentes concentraciones de antígeno.
- Se mide la cantidad de precipitado en cada tubo y se representa en una gráfica, donde la mayor cantidad de precipitado indica la concentración óptima del antígeno.
Técnicas de fijación del complemento
Este procedimiento, aunque sensible, ha sido reemplazado en gran parte por métodos más modernos como ELISA, inmunofluorescencia (IF) y radioinmunoanálisis (RIA). Sin embargo, todavía se utiliza en el diagnóstico de ciertas enfermedades fúngicas, víricas y parasitarias.
El complemento es un conjunto de aproximadamente 20 proteínas plasmáticas, que se activan en cascada tras la interacción entre antígeno y anticuerpo. Cuando esta reacción ocurre sobre células, el complemento puede lisarlas.
Prueba de fijación del complemento
Este método puede utilizarse para detectar antígenos o anticuerpos en una muestra. Se basa en el hecho de que la formación del complejo Ag-Ac bloquea la actividad hemolítica del complemento sobre un sistema de glóbulos rojos sensibilizados.
El procedimiento se divide en dos etapas:
- Reacción primaria
- Se mezclan el antígeno y el anticuerpo en presencia de complemento.
- Si hay una reacción Ag-Ac, el complemento se unirá a esta, evitando que interactúe con otras estructuras.
- Prueba de hemólisis
- Se añaden glóbulos rojos sensibilizados con anticuerpos específicos.
- Se pueden observar dos resultados:
- Ausencia de hemólisis: Indica que el complemento ha sido captado por la reacción Ag-Ac, lo que confirma la presencia del antígeno o anticuerpo buscado.
- Presencia de hemólisis: Sugiere que el complemento sigue libre en la solución, lo que indica que no se formó un complejo Ag-Ac.
Inmunoanálisis con marcadores (Enzimoinmunoensayos, ELISA, inmunofluorescencia, radioinmunoanálisis)
Estos métodos utilizan moléculas indicadoras unidas al antígeno o anticuerpo para demostrar la presencia de una reacción Ag-Ac y detectar la sustancia buscada.
Características principales
- Requieren pequeñas cantidades de reactivos, por lo que son métodos muy eficientes.
- Son rápidos, ya que los resultados pueden obtenerse en minutos u horas (generalmente en menos de 24 horas).
- Poseen alta sensibilidad, lo que permite detectar concentraciones muy bajas de antígenos o anticuerpos.
- Se pueden automatizar, lo que facilita su uso en laboratorios clínicos.
Según el tipo de marcador empleado, se clasifican en:
- Enzimoinmunoanálisis (ELISA o EIA) → Usa enzimas como marcadores.
- Inmunofluorescencia (IF) → Emplea fluorocromos (sustancias fluorescentes).
- Radioinmunoanálisis (RIA) → Utiliza isótopos radiactivos.
Enzimoinmunoanálisis (ELISA)
Este método se basa en la detección de la reacción Ag-Ac mediante el uso de antígenos o anticuerpos marcados con una enzima. La presencia de la enzima en el inmunocomplejo se detecta mediante una reacción de cambio de color al agregar un sustrato específico.
Debido a la actividad enzimática, una sola molécula de enzima puede generar millones de moléculas de producto, lo que confiere a este método una altísima sensibilidad.
Las enzimas más utilizadas como marcadores incluyen:
- Peroxidasa
- Glucosa oxidasa
- Fosfatasa alcalina
La medición del cambio de color se realiza mediante un espectrofotómetro, y en los laboratorios modernos, se emplean lectores de placas automatizados con espectrofotómetros incorporados.
Tipos de ensayos ELISA
Los ELISA pueden dividirse en dos grandes grupos:
- ELISA homogéneo
- La interacción Ag-Ac afecta la actividad de la enzima, lo que permite la detección sin necesidad de separar los componentes libres de los marcados.
- ELISA heterogéneo
- La actividad de la enzima no cambia tras la interacción Ag-Ac, por lo que se requiere una separación física de los inmunocomplejos formados.
- Es el tipo de ELISA más utilizado en la actualidad.
En la mayoría de los casos, estas pruebas se realizan en fase sólida, fijando el antígeno o el anticuerpo en soportes como:
- Placas de microtitulación
- Tubos de plástico
- Perlas de vidrio
Dato clave
El método ELISA permite marcar y detectar un antígeno o anticuerpo con gran precisión, permitiendo su identificación incluso en cantidades extremadamente pequeñas.
Tipos de reacciones de ELISA en fase sólida
1. Método sandwich
Se emplea para detectar antígenos grandes, como los virus de la hepatitis o el gonococo. En este método, el antígeno debe ser polivalente, ya que debe unirse a dos anticuerpos diferentes.
Fases del método sandwich
- Se fija en un soporte el anticuerpo específico del antígeno buscado.
- Se añade la muestra problema (que puede o no contener el antígeno buscado).
- Se incorpora un segundo anticuerpo específico del antígeno, pero marcado con una enzima, permitiendo su detección.
El método descrito, al igual que otro que utiliza un complemento, no es de uso frecuente.
Radioinmunoensayo (RIA)
Estas pruebas permiten detectar el complejo antígeno-anticuerpo (Ag-Ac) mediante el uso de isótopos radiactivos para marcar el antígeno o el anticuerpo.
Los isótopos radiactivos más utilizados incluyen:
- Emisores de radiación beta (β): Carbono 14 (14C), Fósforo 32 (32P) y Tritio (3H).
- Emisores de radiación gamma (γ): Yodo 125 (125I), Yodo 131 (131I) y Cobalto 57 (57Co).
Para detectar la reacción, se emplean contadores de centelleo de rayos gamma o beta, dependiendo del tipo de radioisótopo utilizado. Estos dispositivos miden la radiactividad presente en la muestra.
A pesar de ser pruebas altamente sensibles, presentan ciertas desventajas:
- Se requiere manipulación de radioisótopos, lo que implica riesgos.
- Es necesario seguir estrictas normas de seguridad, lo que limita su aplicación en ciertos laboratorios.
- El equipo necesario es costoso y complejo.
Existen técnicas en las que el antígeno o anticuerpo se inmoviliza en una fase sólida, mientras que en otras se mantiene en fase líquida. Las técnicas más utilizadas corresponden a la primera categoría.
Los métodos del RIA son similares a los del ELISA, predominando los de unión competitiva:
- Marcadores: Se emplean radioisótopos, cuantificables mediante un contador de centelleo.
- Ventajas: Son métodos rápidos, altamente sensibles y relativamente sencillos.
- Desventajas: Requieren instalaciones especializadas y los isótopos tienen una vida media limitada.
Tipos de radioinmunoanálisis:
- Radioinmunoanálisis directo o RIA competitivo: Se utiliza un antígeno marcado que es desplazado por el antígeno no marcado presente en la muestra.
- Radioinmunoanálisis indirecto o RIA no competitivo: Se emplea un anticuerpo antiinmunoglobulina marcado con radiactividad. Un ejemplo de aplicación es la medición de inmunoglobulina E (IgE) específica en individuos con alergias.
Métodos de inmunotransferencia: Western-Blot o Inmunoblotting
Esta técnica se basa en la transferencia de sustancias desde un gel, donde han sido separadas, a un soporte más manejable.
El proceso comienza con la separación de las sustancias en un gel de poliacrilamida mediante electroforesis. Luego, se realiza la transferencia a una membrana, generalmente de nitrocelulosa, mediante capilaridad o electroforesis (electroblotting). Esta membrana tiene la capacidad de fijar proteínas y bloquear sitios de unión no ocupados.
Las sustancias transferidas pueden analizarse con diferentes métodos, principalmente ELISA o RIA, mediante la adición de anticuerpos específicos.
Se trata de una técnica cualitativa, en la que los resultados se observan como bandas que representan cada sustancia presente en la muestra. Actualmente, se usa ampliamente para confirmar la infección por VIH, ya que combina alta sensibilidad con gran especificidad, reduciendo los falsos positivos.
Etapas del proceso:
- Separación del virus VIH desorganizado y parcialmente purificado mediante electroforesis en un gel de poliacrilamida.
- Transferencia de las proteínas separadas a una membrana de nitrocelulosa, donde actuarán como antígenos.
- Adición de la muestra problema (suero con sospecha de anticuerpos contra el VIH) a la membrana.
- Unión de una antiglobulina conjugada con un marcador a los complejos Ag-Ac formados.
- Visualización del resultado mediante ELISA o RIA.
- Comparación de los resultados con un control.
Este método también se usa para la determinación de ácidos nucleicos. Si se analiza ADN, la técnica se denomina Southern blot, mientras que para ARN se conoce como Northern blot.
Otros ensayos inmunológicos
Inmunoturbidimetría e inmunonefelometría
Cuando un haz de luz impacta contra una partícula en suspensión, se producen diversos efectos: una parte de la luz se transmite, otra se absorbe, otra se refleja y otra se dispersa.
Inmunoturbidimetría
Esta técnica mide la luz no transmitida a través de una suspensión cuando se aplica un rayo de luz colimado. La medición se basa en la disminución de la intensidad de la luz incidente debido a dispersión, reflexión y absorción. Se expresa en porcentaje de transmisión o absorbancia.
Cuando un antígeno y un anticuerpo reaccionan en un medio líquido, se generan inmunocomplejos en suspensión que provocan una turbidez proporcional a la cantidad de antígeno o anticuerpo presente en la muestra.
Inmunonefelometría
En este caso, se mide la luz dispersada por las partículas en suspensión cuando se les aplica un haz de luz colimado. La medición se realiza a un ángulo específico (entre 15° y 90°) con respecto al rayo incidente. La formación de inmunocomplejos antígeno-anticuerpo genera una dispersión de la luz que permite detectar la presencia de estos en una muestra.
Ensayos inmunológicos basados en los efectos de la unión Ag-Ac (métodos terciarios)
Estos métodos evalúan los efectos biológicos derivados de la unión antígeno-anticuerpo. Algunos de estos efectos incluyen:
- Fagocitosis:
- Opsonización: Preparación del antígeno para ser fagocitado.
- Adherencia inmunitaria: Favorece la fagocitosis.
- Desgranulación celular.
- Bacteriólisis.
- Inmovilización de microorganismos.
La mayoría de estos efectos están asociados a la actividad del complemento. Algunas de las pruebas más relevantes son:
Pruebas de protección
Estas pruebas evalúan la presencia de anticuerpos protectores responsables de la inmunidad adquirida.
- Activas: Se administra un antígeno a un grupo de animales y, tras un tiempo determinado, se les inocula un germen virulento. La protección se mide en función de la supervivencia.
- Pasivas: Se administra un suero con anticuerpos a un grupo de animales y luego se les inocula un germen virulento. La protección se evalúa de manera similar.
Pruebas de neutralización
Cuando un antígeno se une a su anticuerpo específico, puede perder su capacidad patógena, tóxica, hormonal o enzimática. Para demostrarlo, se inocula la mezcla Ag-Ac en un animal de experimentación o en un cultivo. Por ejemplo, al combinar una toxina con su antitoxina, se neutraliza su efecto tóxico.
Pruebas de hipersensibilidad
Estas pruebas permiten evaluar la sensibilidad de un individuo a sustancias como medicamentos, pólenes, productos químicos, polvo o alimentos.
- Test cutáneos intradérmicos: Se inyecta una sustancia bajo la piel y se observa la reacción.
- Parches cutáneos: Se escarifica la piel y se coloca un parche con la sustancia a evaluar.
- Pruebas de provocación: Se administran alérgenos por vía natural (inhalación, vía oral, etc.) para observar reacciones.
- Test de Prausnitz-Küstner: Se inyecta suero de un paciente alérgico en un individuo sano y se expone al alérgeno tras 24 horas.
- Prueba de tuberculina: Se inyecta tuberculina en la piel y se mide la reacción tras 48-72 horas.
Estudio de las Diferentes Poblaciones Linfocitarias
En los métodos inmunológicos, no solo se debe analizar la reacción antígeno-anticuerpo y sus efectos, sino también estudiar las células involucradas en la inmunidad celular, particularmente los linfocitos T y B.
Existen técnicas que permiten aislar los linfocitos del resto de los componentes sanguíneos, basadas en la velocidad de sedimentación o la centrifugación en gradiente de densidad. Además, hay métodos para identificar distintas subpoblaciones linfocitarias, como la prueba de formación de rosetas E, que aprovecha la capacidad de los linfocitos T para formar rosetas espontáneamente con los glóbulos rojos de carnero, sin intervención de inmunoglobulinas.
En algunos casos, es necesario evaluar la capacidad funcional de los linfocitos, lo que resulta más útil para determinar el estado inmunológico de un individuo que simplemente cuantificarlos. Se pueden medir la producción de anticuerpos y la respuesta a mitógenos. Entre las técnicas más relevantes están la transformación linfoblástica, el cultivo mixto de linfocitos y la proliferación de linfocitos en respuesta a diversas sustancias, como las lecitinas vegetales.
Quimioluminiscencia
Definición
La luminiscencia es la emisión de luz provocada por la disipación de energía en una sustancia que ha sido excitada electrónicamente. La quimioluminiscencia ocurre cuando una molécula libera un fotón como resultado de una reacción química en la que uno de los productos intermedios o finales alcanza un estado excitado y, al regresar a su estado fundamental, emite luz. Se puede definir como la generación de luz mediante reacciones químicas. La mayoría de estas reacciones son oxidativas, ya que requieren una alta cantidad de energía para producir un fotón y suelen involucrar oxígeno o peróxido de hidrógeno como sustratos oxidables.
Tipos de Luminiscencia
Existen diversas formas de luminiscencia, diferenciadas por el mecanismo que las produce:
- Fotoluminiscencia: También conocida como fluorescencia, ocurre cuando una sustancia es estimulada por fotones de luz, generando una emisión de luz con un trazador fluorescente.
- Bioluminiscencia: Se genera a través de una reacción química mediada por enzimas y está relacionada con organismos vivos, como en el caso de las luciérnagas.
- Quimioluminiscencia: En este proceso, la emisión de luz es causada por los productos de una reacción química específica. Dependiendo del sistema automatizado utilizado, pueden intervenir sustancias como el éster de acridina, peróxido-ácido, hidróxido de sodio y fosfatasa alcalina. En esta reacción, el éster de acridina es oxidado por el peróxido-ácido y el hidróxido de sodio.
Fundamento de la Quimioluminiscencia (CLIA)
La cuantificación de una sustancia mediante quimioluminiscencia se basa en la reacción antígeno-anticuerpo, utilizando un marcador indicador como el éster de acridina u otro compuesto similar. En combinación con los reactivos peróxido-ácido e hidróxido de sodio, se desencadena una reacción quimioluminiscente cuando la muestra es analizada.
El peróxido-ácido actúa como agente oxidante del éster de acridina, mientras que el hidróxido de sodio ajusta el pH para facilitar la oxidación. La combinación de una enzima con una reacción de detección basada en quimioluminiscencia o bioluminiscencia permite una alta sensibilidad analítica.
Durante el proceso, el antígeno en la muestra del paciente compite con partículas paramagnéticas unidas covalentemente a un anticuerpo marcado con éster de acridina. Existe una relación inversa entre la concentración de anticuerpo marcado y la cantidad de antígeno presente en la muestra del paciente.
El ensayo CLIA es de tipo sándwich, en el que el antígeno de la muestra se somete a reacción con un anticuerpo unido covalentemente a partículas paramagnéticas y otro anticuerpo marcado con éster de acridina. La concentración de antígeno en la muestra del paciente está directamente relacionada con la cantidad de luz emitida durante la oxidación del éster de acridina en la cubeta.
Para cuantificar el antígeno, el sistema automatizado inyecta dos reactivos en las cubetas con la mezcla de reacción, lo que inicia la emisión de fotones de luz. Un fotomultiplicador (PMT) detecta los fotones y los convierte en pulsos eléctricos, los cuales son contados y comparados con una curva maestra para calcular la concentración de la sustancia analizada.
Aplicaciones de la Quimioluminiscencia
Los análisis realizados con esta técnica incluyen:
- Perfil tiroideo
- Perfil ginecológico
- TSH
- BHGC
- T4 total y T4 libre
- T3 total
- LH y FSH
- Prolactina
- Progesterona
- Estradiol
- Diagnóstico de alergias
- Marcadores metabólicos
- Inmunoglobulina E (IgE)
- Cortisol
- Perfil de anemia
- Diagnóstico de enfermedades infecciosas
- Vitamina B12
- Toxoplasma (IgG e IgM)
- Ferritina y rubéola (IgG e IgM)
- Folatos
- Chlamydia Ag
- Virus sanguíneos
- Detección de drogas terapéuticas
- VIH 1+2
- Teofilina (tercera generación)
- Antígeno de superficie de hepatitis B (HBs Ag)
- Prueba confirmatoria de HBs Ag
- Digoxina
- Diagnóstico de diabetes y enfermedades cardiovasculares
- Insulina
- CK-MB
- Mioglobina
- Troponina I
Esta técnica proporciona una alta sensibilidad y especificidad en el análisis clínico, facilitando el diagnóstico de diversas enfermedades y condiciones médicas.
Química Seca
Fundamento
Cuando una superficie es expuesta a radiación, pueden ocurrir dos fenómenos:
- Reflexión especular: Se produce en superficies lisas y pulidas, donde la radiación se refleja de manera uniforme, manteniendo el ángulo de incidencia igual al de reflexión.
- Reflexión difusa: Tiene lugar en superficies rugosas o irregulares, dispersando la radiación en múltiples direcciones. La fotometría de reflectancia se basa en este tipo de reflexión, midiendo la energía reflejada en la longitud de onda que absorbe el analito.
La relación entre la reflectancia (R) y la concentración del analito no es lineal. Un objeto negro absorbe toda la radiación (R = 0), mientras que un objeto blanco la refleja completamente (R = 1).
Instrumentación
La fuente de radiación más común es una lámpara de Wolframio. Se utilizan filtros como selectores de longitud de onda y los detectores empleados son similares a los usados en el espectro visible.
Aplicaciones
Esta técnica se emplea en análisis de orina mediante tiras reactivas. Cada tira contiene varias secciones impregnadas con reactivos diferentes, permitiendo realizar múltiples determinaciones simultáneamente. Al entrar en contacto con la orina, los reactivos reaccionan con los compuestos presentes en la muestra. Por ejemplo, en la sección destinada a la glucosa, la tira está impregnada con reactivos como KL, glucosa oxidasa (GOD) y un tampón, lo que da lugar a una reacción química específica que genera un cambio de color.
El equipo cuenta con distintos filtros para medir la reflectancia del compuesto resultante, facilitando la identificación de sustancias presentes en la muestra.
Medida de Reflectancia en Placa
En este método, los reactivos están contenidos en una placa o “slide”, donde se aplica la muestra y se desarrolla la reacción química. Algunos sistemas, como los empleados en análisis bioquímicos de Kodak, utilizan este formato.
Turbidimetría
Fundamento
Cuando un haz de radiación interactúa con partículas en suspensión, puede sufrir procesos como adsorción, reflexión o dispersión. La concentración de partículas en suspensión se puede determinar de dos maneras:
- Turbidimetría: Mide la disminución de la intensidad de la luz incidente debido a la presencia de partículas en suspensión.
- Nefelometría: Evalúa la luz dispersada en un ángulo específico.
Turbidimetría
La turbidimetría se basa en la medición de la transmisión de luz que es bloqueada por partículas en suspensión. Cuantifica la luz residual transmitida y, si la muestra es coloreada, se utilizan filtros para evitar interferencias con la absorción de la radiación.
Instrumentación
Similar a la espectroscopia de absorción visible, consta de una fuente de radiación, un filtro, una cubeta con la muestra y un detector.
Aplicaciones
Esta técnica es ampliamente utilizada en bacteriología para evaluar la sensibilidad a antibióticos, en analizadores de coagulación, en la determinación de proteínas totales y en muestras de orina y líquido cefalorraquídeo.
Nefelometría
Fundamento
La nefelometría mide la luz dispersada por partículas en suspensión, colocando un detector en un ángulo específico respecto a la fuente de radiación. La intensidad de la luz dispersada está relacionada con la concentración de partículas y depende de su tamaño, cantidad e índice de refracción. Es una técnica más sensible que la turbidimetría, por lo que se prefiere en análisis donde las concentraciones a medir son bajas.
Instrumentación
La mayoría de los nefelómetros detectan la luz dispersada a 90°, ya que esta configuración es más fácil de fabricar. Sin embargo, teóricamente, se obtiene mayor sensibilidad en ángulos más pequeños.
Aplicaciones
Se usa principalmente en la detección de reacciones antígeno-anticuerpo, ya que los complejos formados son capaces de dispersar la luz. Ejemplos de aplicación incluyen la determinación de inmunoglobulinas, apoproteínas y fracciones del complemento.
Resumen
La nefelometría es un método analítico basado en la dispersión de luz en presencia de partículas en suspensión. Cuando un haz de luz atraviesa un medio transparente que contiene partículas sólidas, se dispersa en múltiples direcciones, generando un aspecto turbio en la muestra. Este principio se aplica en múltiples áreas de análisis clínico y de laboratorio.
Refractometría de Líquidos
Fundamento
Cuando la luz pasa de un medio a otro con diferente densidad, su dirección de propagación cambia debido a la variación en la velocidad de la radiación en cada medio. Este fenómeno se conoce como refracción y ocurre cuando la luz atraviesa una sustancia ópticamente transparente.
El índice de refracción (n) mide el grado de refracción o cambio de dirección de la luz, dependiendo del ángulo de incidencia y el ángulo de refracción. Se expresa como:
- n = 1: Índice de refracción del aire.
- n: Índice de refracción de la sustancia ópticamente transparente.
Factores que influyen en el índice de refracción:
- Temperatura.
- Propiedades del líquido.
- Longitud de onda de la radiación incidente.
- Concentración.
Si se mantienen constantes los tres primeros factores, el índice de refracción puede relacionarse directamente con la concentración, lo que constituye la base de la refractometría.
Instrumentación
La medición se basa en el ángulo límite y se lleva a cabo con dispositivos formados por prismas y lentes que dirigen la luz a un ángulo determinado. Uno de los más empleados es el Refractómetro de Abbe.
Aplicaciones
Se utiliza para determinar la concentración de proteínas totales y para el cálculo de la densidad de la orina.
Fotometría de Reflectancia
Fundamento
Cuando una superficie es iluminada por radiación, pueden ocurrir dos tipos de reflexión:
- Reflexión especular: Se presenta en superficies lisas y pulidas, donde la radiación se refleja en un solo ángulo, igual al de incidencia.
- Reflexión difusa: Se produce en superficies rugosas o no pulidas, donde la radiación se dispersa en múltiples direcciones. Este principio es la base de la fotometría de reflectancia, que mide la energía reflejada de manera difusa a una longitud de onda específica absorbida por el analito.
La relación entre la reflectancia (R) y la concentración del analito no es lineal. En términos generales:
- Una superficie negra absorbe toda la radiación, resultando en una reflectancia de 0.
- Una superficie blanca refleja toda la radiación, alcanzando una reflectancia de 1.
Instrumentación
La fuente de radiación suele ser una lámpara de Wolframio. Para seleccionar la longitud de onda adecuada, se utilizan filtros, y los detectores empleados son similares a los utilizados en el espectro visible.
Aplicaciones
Se usa en análisis de orina mediante métodos de tira reactiva. Estas tiras contienen múltiples zonas impregnadas con distintos reactivos, permitiendo realizar varias pruebas simultáneamente.
Por ejemplo, en la determinación de glucosa, una sección de la tira contiene reactivos como KL, glucosa oxidasa (GOD) y un tampón, que adquiere un color azul. Cuando entra en contacto con orina que contiene glucosa, ocurre la siguiente reacción:
Glucosa + H₂O + O₂ → Ácido glucónico + H₂O₂
H₂O₂ + 3 KL + 2 H⁺ → KL + 2K⁺ + 2H₂O
El equipo de medición emplea distintos filtros para seleccionar la longitud de onda adecuada y determinar la reflectancia del compuesto resultante, generalmente de color marrón.
Medición de Reflectancia en Formato Placa
En este método, los reactivos están depositados en una placa o “slide”, donde se añade la muestra y se desencadena la reacción. Algunos sistemas, como los utilizados en los análisis bioquímicos de Kodak, emplean este formato para mejorar la precisión en la medición de la reflectancia.
